
Abstract
Using the OSMAC (One Strain MAny Compounds) approach, the fungal endophyte Fusarium tricinctum was cultivated on fruit and vegetable juice-supplemented solid rice media. This led to an up to 80-fold increase in the accumulation of the new natural product fusarielin J (1), as well as to the induction of two new natural products fusarielin K (2) and fusarielin L (3) and the known derivatives fusarielins A (4) and B (5). Compounds 2–5 were not detected when the fungus was grown on rice media lacking either fruit or vegetable juice. The highest increase in the accumulation of compound 1 was observed in the presence of apple and carrot juice, whereas the stimulating effect was weaker for banana juice. Compound 1 exhibited cytotoxicity against the human ovarian cancer cell line A2780, with an IC50 value of 12.5 μM.
Similar content being viewed by others
Introduction
The fungal genus Fusarium is most prominently known for its phytopathogenic effects on various staple food crops, causing depletory ‘Fusarium wilt’ (Panama Disease) in bananas1 and ‘Fusarium head blight’ in cereals, such as wheat, oat, barley and maize.2 Fusarium species (for example, F. graminearum and F. tricinctum) produce mycotoxins that are potentially harmful to humans, whereas some of them can also be used as drugs or drug candidates. These mycotoxins include beauvericins, trichothecenes and most importantly enniatins3, 4 that were medicinally used (fusafungine) up to the beginning of this year as local antibiotics in nasopharyngitis treatment.5 Enniatins are currently being investigated as potential anticancer agents,6, 7 and so are semisynthetic derivatives of the cyclic depsipeptide sansalvamide A, another Fusarium metabolite with strong in vitro cytotoxicity against pancreatic, colon and breast cancer cell lines.8, 9, 10, 11 In addition, Fusarium strains have transiently produced several antineoplastic agents formerly known from plants such as paclitaxel, camptothecin and L-asparaginase,12, 13, 14 as well as podophyllotoxin,15 the latter being the lead compound that led to the clinically used semisynthetic drugs etoposide and teniposide.
These examples demonstrate how auspicious secondary metabolites from Fusarium species are and that ongoing natural product research on this genus promises to be a highly rewarding strategy for the discovery of new bioactive compounds.16
One of the approaches for drug discovery from fungi involves the application of the OSMAC (One Strain MAny Compounds) method that attempts to induce silent biogenetic clusters and hence may lead to accumulation of compounds not accumulated during more conventional fermentations. OSMAC terms the modulating effect that altered culture conditions (that is, media composition, temperature, osmolarity, pH) may have on the secondary metabolite production of microorganisms.17, 18 Examples for such culture variations for filamentous fungi include the use of different liquid or solid media, as applied in a previous study with F. tricinctum fermented on solid beans and liquid Wickerham medium versus cultivation on solid rice medium, leading to an enhanced production of enniatins on beans.19, 20 Another example is the mimicry of extreme habitats by cultivating at colder temperatures or using highly saline media (for example, growing Gymnascella dankaliensis on either NaCl- or NaBr-enriched rice medium) that led to the induction of chlorinated or brominated metabolites.21, 22, 23
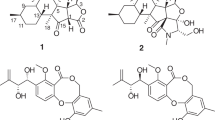
With Fusarium species such as F. graminearum and F. tricinctum, addition of CaBr2 to the cultivation medium led to an induced production of the bioactive compound fusarielin A.24, 25 Fusarielin-type polyketides are a therapeutically promising class of Fusarium metabolites: with 17 known derivatives identified so far (fusarielin A–I, ICM0301 A–H),26, 27, 28, 29, 30 fusarielins display antifungal,28 mild antibacterial24, 31 and anti-angiogenic activities,27, 32 in addition to weak cytotoxicity against HeLaS3 and NCI-H69 cells.26 Interestingly, previous cultivation experiments have shown enhanced fusarielin yields by cultivating F. tricinctum on complex carbon sources.25 Therefore, having an endophytic F. tricinctum strain at hand that was previously isolated from the rhizome of Aristolochia paucinervis, we decided to implement the OSMAC concept by cultivating it on solid rice medium supplemented with three types of commercially available fruit and vegetable juices, namely apple, banana and carrot juice, as these naturally contain a high percentage of various carbohydrates. Herein, cultivating F. tricinctum on rice containing juice had a starkly enhancing and inducing effect on the production of fusarielins: the new fusarielin J (1) was enhanced 80-fold compared with control cultures lacking juice and the production of the new fusarielins K (2) and L (3) and the known fusarielins A (4) and B (5) was induced. A total of 14 natural products were identified based on their NMR and MS spectra and all fusarielins (Figure 1) were investigated for their cytotoxicity against A2780 human ovarian cells.

Structures of compounds 1–5.
Results and Discussion
F. tricinctum was cultivated on solid rice medium with or without fruit or vegetable juice in two flasks per medium variation. In each case the fermentation was stopped by addition of ethyl acetate after 14 days and all experiments were carried out in triplicate, resulting in 6 cultivation flasks per medium.
The ethyl acetate extracts of all treatments presented typical metabolic profiles in HPLC analysis (Figure 2): the known compounds (−)-citreoisocoumarin (6), (−)-citreoisocoumarinol (7), lateropyrone (8), enniatin B (9), enniatin B1 (10), enniatin A1 (11), enniatin A (12) and fusaristatin A (13) were readily identified by LC-MS analysis and by comparison with authentic standards that had either previously been isolated from the same fungal strain,33 or in the case of phenyl acetic acid (14) by comparison with a commercially available standard. The amounts of 6–13 per flask irrespective of the presence or absence of juice were comparable to those of previous experiments (Table 1).33 Compound 14 was isolated from extracts of juice-supplemented cultures; however, it was not detectable in the HPLC chromatograms. Interestingly, the juice-based cultivations enhanced the production of the new natural product fusarielin J (1) up to 80-fold compared with fungal cultures lacking juice (Table 1). Furthermore, the cultivation of F. tricinctum with juice induced the production of two new natural products fusarielin K (2) and fusarielin L (3), and of two known natural products fusarielins A (4) and B (5) that were not detected in fungal controls. None of the investigated compounds including the new natural products 1–3 were detected when only the juices used for the described experiments were analyzed.

HPLC chromatograms (min vs mAU) overlay of EtOAc extracts: Fusarium tricinctum on solid rice medium prepared with water (a), banana (b), apple (c) and carrot juice (d); compound 14 was not detectable. A full color version of this figure is available at the Journal of Antibiotics journal online.
Compound 1 was isolated as a white, amorphous solid and exhibited a UV absorption maximum at 241 nm. The high-resolution electrospray ionization mass spectrometry (HRESIMS) of 1 displayed a prominent ion peak at m/z 561.3182 [M+Na]+ indicating the molecular formula C33H46O6, consistent with 11 degrees of unsaturation. In the 1H NMR spectrum of 1, 43 protons were detected, including two pairs of overlapping aromatic protons at δH 7.26 (brd, 7.4, H-4′/8′) and 7.34 (brt, 7.4, H-5′/7′), one aromatic proton at δH 7.30 (brt, 7.4, H-6′), four olefinic protons at δH 6.18 (dd, J=15.0, 10.8, H-6), 5.85 (dd, J=10.8, 1.2, H-5), 5.29 (qd, J=6.7, 1.5, H-19) and 5.02 (dd, J=15.0, 10.2, H-7), five downfield shifted aliphatic protons at δH 4.70 (brq, J=2.2, H-11), 3.78 (d, J=9.0, H-3), 3.55 (dd, J=10.6, 5.8, Hα-1), 3.70 (dd, J=10.6, 5.0, Hβ-1) and 2.74 (brs, H-15), suggesting their direct vicinity to an oxygen atom, three methylene groups at δH 1.53 (t, J=14.0, Hα-13) and 1.65 (m, Hβ-13), 1.27 (m, Hα-10) and 1.40 (m, Hβ-10), and 3.62 (s, H2-2′), five methine protons at δH 2.55 (d, J=5.2, H-17), 2.18 (m, H-8), 1.91 (ddd, J=14.0, 11.1, 3.0, H-14), 1.80 (m, H-2) and 1.55 (m, H-9), and six methyl groups at δH 1.69 (d, J=1.2, H3-22), 1.67 (d, J=6.7, H3-20), 1.61 (brs, H3-25), 1.21 (s, H3-24), 1.04 (s, H3-23) and 0.78 (d, J=6.9, H3-21). The COSY spectrum revealed four spin systems that consisted of H-21/H-2/H3-21/H-3 (A), H3-22/H-5/H-6/H-7/H-8/H-17/H-9/H2-10/H-11/H2-13/H-14 (B), H-19/H3-20/H3-25 (C) and H-4′/H-5′/H-6′/H-7′/H-8′ (D) (Figure 3a). After assigning all protons to their attaching carbon atoms via analysis of the HSQC spectrum (Table 2), the aforementioned spin systems were connected through interpretation of the heteronuclear multiple bond correlation (HMBC) spectrum as follows: the first spin system (A) was connected to the second (B) by the correlation from H-3 to the quaternary carbon C-4 (δC 137.4) and CH3-22 (δC 11.5), and by the J3 correlation from H-5 to the oxygenated C-3 (δC 81.9). The third spin system (C) was elucidated as a but-2-ene moiety through a J2 correlation from H3-25 to the quaternary carbon C-18 (δC 134.7) and J3 correlations to C-19 (δC 126.1). Moreover, the HMBC correlation from H3-25 to C-17 and from H-8 to C-18 connected this moiety (C) to the decalin system at position C-17. By further HMBC evaluation and comparison with the known fusarielin B (5), which was also isolated in this study, the basic framework of 1 was found to be identical to that of 5.26

(a) Key COSY correlation (bold lines) and heteronuclear multiple bond correlation (HMBC; arrows) of 1; (b) key HMBC (plain arrows) and NOE (dashed arrows) correlations of the 2,2,5-trimethyl-1,3-dioxane moiety of 2. A full color version of this figure is available at the Journal of Antibiotics journal online.
The remaining signals (spin system D and H2-2′) were attributed to a phenyl acetate unit, as supported by the HMBC correlations from the aromatic protons H-4′, H-5′, H-7′ and H-8′ to C-3′ (δC 135.7) and the methylene carbon C-2′ (δC 42.7) and from H-6′ to C-3′. Similarly, the methylene protons H2-2′ correlated with C-3′ as well as with the aromatic carbons C-4′/8′ and C-5′/C7′ (δC 129.4) and the carbonyl carbon C-1′ (δC 171.8). The connection of the phenyl acetate function to the fusarielin B (5) framework was established through the HMBC correlation from H-11 to C-1′ (Figure 3a), and was corroborated by the marked downfield shift (Δ 1.43 p.p.m.) of this proton, as a result of deshielding effects from the resulting ester function. The relative configuration of 1 was deduced by the J-values extracted from the 1H NMR spectrum (Table 2) and by interpretation of the 2D NOESY spectrum (Figure 3a) to be identical to that of fusarielin B (5). Thus, compound 1 was identified as a new natural product and was named fusarielin J.
Compound 2 was isolated as a white, amorphous solid and showed a similar UV–visible spectrum as 1. The molecular formula C36H50O6 was deduced by the prominent ion peak at m/z 596.3944 [M+NH4]+ in the HRESIMS, suggesting an additional structural functionality of 40 amu compared with 1. The 1H NMR spectrum of 2 was very similar to that of 1, indicating that both compounds shared the same fusarielin core structure. Additional signals were detected at δH 1.34 (s, H3-26) and 1.48 (s, H3-27), assigned to two methyl groups. In the HMBC spectrum of 2, the correlations of both CH3-26 and CH3-27 with each other as well as with the remaining ketal quaternary carbon C-1′′ (δC 99.3) revealed the presence of an additional acetonide moiety. The HMBC correlation from H2-1 to C-1′′ confirmed the position of the acetonide group through ether linkages at C-1 and C-3 (Figure 3b), rationalizing the additional degree of unsaturation. The relative configuration of the fusarielin core structure of 2 was determined to be identical to that of 1 on the basis of the respective coupling constants and NOESY correlations (Table 3). In addition, the six-membered dioxane ring adopted a chair conformation (Figure 3b), as suggested by the NOE correlations of H3-27 with the axial protons H-1α and H-3, indicating their co-facial orientation. This was further corroborated by the 13C chemical shifts of CH3-26 and CH3-27 (δC 29.9 and 19.4, respectively) that are characteristic of a syn 1,3-diol acetonide moiety. The acetonide function is known from other fungal natural products; for example, ganodermacetal from the basidiomycete Ganoderma amboinense,34 or cochliomycins A and B from the ascomycete Cochliobolus lunatus.35 The isolation of 2 was performed in the absence of acetone, and the fact that no acetonide was formed, even after incubating 1 mg of 2 in 1 ml acetone for 24 h strongly suggested that 2 is a natural product and not an artifact arising from 1 during the isolation procedure. Therefore, compound 2 was identified as a new natural product and was given the name fusarielin K.
Compound 3 was isolated as a colorless, amorphous solid and displayed a similar UV spectrum as 1 and 2. The molecular formula of 3 was identified as C28H42O4 based on its prominent ion peaks detected at m/z 465.2978 [M+Na]+, revealing a 40 amu increase in the molecular weight compared with fusarielin A (4). The 1H and 13C NMR data of 3 were similar to those of 4, apart from the presence of two additional methyl signals at δH 1.33 (s, H3-26) and 1.46 (s, H3-27), thus suggesting the presence of an additional acetonide moiety as found in 2. This assumption was further corroborated by careful analysis of the HMBC spectrum of 3, in which the cross peaks from Hβ-1 (δH 3.72), H3-26, and H3-27 to C-1′ (δC 99.4) were discerned. The relative configuration of 3 was established on the basis of NOE correlations, coupling constants and 13C NMR data (Table 4), by analogy with 1 and 2. Therefore, 3 was identified as the 1,3-diol acetonide derivative of 4 and was given the name fusarielin L.
The known compounds fusarielins A (4) and B (5) were identified by comparison of their spectroscopic data with those in the literature.26 In addition, the absolute configurations of 4 and 5 (Figure 1) had previously been deduced on the basis of CD spectroscopy and chemical derivatization experiments.26 Therefore, based on the similar NMR data and specific optical rotation values of 1, 2 and 3 ([α]20D -76, -53 and -33, respectively) compared with those of 4 and 5 ([α]20D -132 and -38, respectively) as well as on their close biogenetic relationship, it is assumed that these compounds (1–5) share the same absolute configuration (Figure 1).
In a previous study on F. tricinctum, the influence of different carbon and nitrogen sources, as well as variation of pH, cultivation time and temperature had been analyzed.25 Interestingly, the use of complex carbon sources showed the strongest enhancement of fusarielin A (4) production.25 As juices are naturally high in carbohydrates (6–14 g per 100 ml), our experiments clearly underpin these findings. Moreover, our juice experiments led to an additional enhancement and/or induction of more and new fusarielin derivatives (1–3). However, the carbohydrate content (banana>apple>carrot) of each juice was not coherent with the yield of fusarielins per flask (apple>carrot>banana) (Table 1). Therefore, other juice components/factors may play an additional role in fusarielin induction and enhancement. In order to pinpoint which physical characteristic(s) of the juices (also known as OSMAC effector) comports linear to the induction and enhancement of fusarielins, we compared several properties of the juices and media, including pH, osmolarity and juice contents, but no interrelations were found (Table 5). This is not surprising as previous OSMAC studies (for example, using tap water versus distilled water) revealed that even slight changes in the cultivation medium, such as differences in metal ion concentrations, trigger the production of new bioactive secondary metabolites in endophytic fungi.36
All fusarielins (1–5) were submitted to a cytotoxicity assay against the human ovarian cancer cell line A2780. Compound 1 exhibited moderate cytotoxicity with an IC50 value of 12.5 μM against the A2780 cells. All other fusarielins (2–5) showed very weak activity (>36 μM) in the same assay. For the known fusarielins A (4) and B (5), this was in accordance with previously published data.26 Interestingly, the acetonide function in fusarielin K (2) and L (3) led to a three- and twofold decreased activity compared with the precursors fusarielins J (1) and A (4) (Table 6). The phenyl acetic acid function seems to play an important role in the cytotoxic activity of fusarielin J (1) and K (2). Compound 1 is fourfold more cytotoxic than phenyl acetic acid lacking fusarielin A (4) and B (5), and 2 is about twofold more cytotoxic than fusarielin L (3).
To the best of our knowledge, this is the first report using fruit or vegetable juices for an OSMAC approach, and from the fusarielin derivatives identified in this study so far only fusarielin A (4) had been isolated from cultures of F. tricinctum before.24, 25, 37 In addition, we were able to induce the production of the known fusarielins A (4) and B (5), and of two new fusarielins K (2) and L (3), along with the 80-fold enhancement of the new fusarielin J (1). In conclusion, employing juice as an addition to the fermentation media of fungi proves to be a promising OSMAC approach for the discovery of new bioactive secondary metabolites in the ongoing quest of natural product research to provide model structures for the development of potential future drugs.
Experimental Procedure
General experimental procedures
For a measurement of OR of chiral compounds, the latter were dissolved in optically pure solvents (Uvasol, Merck Chemicals, Darmstadt, Germany) and left to equilibrate for 30 min. The specific OR was then measured thrice and averaged for each sample, using a PerkinElmer 241MC polarimeter (Perkin-Elmer, Waltham, MA, USA). All NMR data (1D and 2D) were measured in deuterated methanol on Bruker Avance III 300, Bruker Avance DRX 500 or Bruker Avance III 600 MHz NMR spectrometers at 25 °C (Bruker BioSpin, Rheinstetten, Germany), and calibrated with the deuterated methanol peak (δH 3.31 p.p.m. and δC 49.0 p.p.m.). The molecular weight of each compound was determined using an HP110 Agilent Finnigan LCQDecaXP Thermoquest ESI mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), and the molecular formula was assigned with the help of HRESIMS spectra measured on a UHR-TOF maxis 4G mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). All fractions and pure compounds were monitored via analytical HPLC DIONEX p580 UVD340S PDA DAD3000RS (Dionex Softron, Munich, Germany).38 Compounds were purified via semipreparative HPLC Merck Hitachi HPLC, I-7100 pump and I-7400 detector (Merck KGaA, Darmstadt, Germany)38 or preparative TLC polygramSIL G/UV254, pre-coated (Macherey-Nagel, Düren, Germany), using HPLC grade eluents (Merck Chemicals) in each case. Initial and subsequent fractionation of crude extracts was performed using column chromatography with stationary phase either silica gel 60M (Macherey-Nagel)38 or Sephadex LH-20 25–100 μm bead size (Sigma-Aldrich, Munich, Germany). TLC plates were used to monitor the collected fractions by spotting them at 254 and 366 nm. The osmolarity of the juices was measured with an osmomat3000 osmometer (Gonotec, Berlin, Germany). The pH values of the rice media were determined using pH-indicator strips (Merck Chemicals).
Microbial material
F. tricinctum had previously been isolated as an endophyte from rhizomes of Aristolochia paucinervis collected in Beni-Mellal (Morocco).33 Fresh mycelia of F. tricinctum were grown on agar plates39 and used for all cultivation experiments.
Culture conditions
Aqueous solid rice culture control medium was prepared following our standard operation protocol.39 For the OSMAC study, the demineralized water in the mentioned protocol was replaced with banana juice (Pfanner, St Peter, Germany), apple juice (Naturkind, Kaiser’s Tengelmann, Mülheim an der Ruhr, Germany) and carrot juice (Naturkind) by volume measurement (110 ml per 100 g rice, autoclavation at 121 °C for 20 min). Each medium was prepared in duplicate, resulting in eight Erlenmeyer flasks in total per experiment that was performed three times. Per flask, half an agar plate mycelium was added under sterile conditions, using four fully overgrown plates per experiment in total. The cultures were left to grow at room temperature (21 °C) and in the dark. After 14 days, the mycelia of F. tricinctum had completely covered and grown through each medium, and thus were harvested at this time. This experiment was repeated twice.
Extraction and isolation
Each flask was infused with 600 ml EtOAc, the solid medium was chopped up inside the flask and after covering each flask up, they were extracted on a horizontal shaker (140 r.p.m.) for 8 h. Subsequently the flasks were left in a static state overnight before being filtered and dried over a rotary evaporator to generate the EtOAc crude extract per flask. The mean dry weight yield of the aqueous cultures amounted to ca. 180 mg per flask, of the banana juice cultures to ca. 275 mg per flask, of the apple juice cultures to ca. 307 mg per flask and of the carrot juice cultures to ca. 353 mg per flask. For initial characterization, each extract was dissolved in 50 ml MeOH (HPLC grade, VWR, Radnor, PA, USA) and injected into the above-mentioned analytical HPLC with 20 μl per extract. The extracts of the apple and carrot juice cultures were combined and then submitted to vacuum LC separation. As these yielded a sufficient quantity, extracts from banana juice were not included. The fraction obtained with the eluent n-hexane/EtOAc (1:4) amounted to 290 mg and was further separated over a Sephadex column with methanol. Five of the so generated subfractions were further worked on: fraction 3 (96 mg) was purified via preparative TLC (methanol/DCM, 1:4) yielding compounds 1 (18.8 mg), 4 (5.2 mg), 2 (3.3 mg), 5 (7.2 mg) and 3 (1.4 mg).
Compounds 7–14 were identified from the crude extracts via LC-MS analysis with the corresponding known compounds previously isolated in our group.33 An experimental calibration curve (concentration vs area units; R2 >0.9) was produced for each purified compound by HPLC measurements of several concentrations, so that the yields per flasks for each compound could be calculated from the resulting curve function.
Fusarielin J (1)
White, amorphous, hygroscopic solid; [α]20D -76° (c 0.14, MeOH); UV (MeOH) λmax (log ɛ) nm 243 (14.7); 1H, 13C and 2D NMR data Table 2; HRESIMS m/z 561.3182 [M+Na]+ (calcd for C33H46O6, 561.3184).
Fusarielin K (2)
White, amorphous, hygroscopic solid; [α]20D -53° (c 0.14, MeOH); UV (MeOH) λmax (log ɛ) nm 243 (14.1); 1H, 13C and 2D NMR data Table 3; HRESIMS m/z 596.3944 [M+NH4]+ (calcd for C36H54NO6, 596.3946).
Fusarielin L (3)
Colorless solid; [α]20D -33° (c 0.11, MeOH); UV (MeOH) λmax (log ɛ) nm 242 (15.0); 1H, 13C and 2D NMR data Table 4; HRESIMS m/z 465.2978 [M+Na]+ (calcd for C28H42O4Na, 465.2975).
Biological assays
All fusarielins (1–5) isolated in this study were investigated in an MTT assay against the cisplatin-sensitive A2780 human ovarian cancer cell line, following previous protocols.40, 41
References
Ploetz, R. C. Management of Fusarium wilt of banana: a review with special reference to tropical race 4. Crop. Prot. 73, 7–15 (2015).
Kim, S. H. & Vujanovic, V. Relationship between mycoparasites lifestyles and biocontrol behaviors against Fusarium spp. and mycotoxins production. Appl. Microbiol. Biotechnol. 100, 5257–5272 (2016).
Kokkonen, M., Ojala, L., Parikka, P. & Jestoi, M. Mycotoxin production of selected Fusarium species at different culture conditions. Int. J. Food Microbiol. 143, 17–25 (2010).
Ola, A. R. B., Aly, A. H., Lin, W. H., Wray, V. & Debbab, A. Structural revision and absolute configuration of lateritin. Tetrahedron Lett. 55, 6184–6187 (2014).
CMDh CMDh endorses revocation of authorisations for fusafungine sprays to treat airway infections, 227560 (European Medicines Agency, (2016).
Wätjen, W. et al. Enniatins A1, B and B1 from an endophytic strain of Fusarium tricinctum induce apoptotic cell death in H4IIE hepatoma cells accompanied by inhibition of ERK phosphorylation. Mol. Nutr. Food. Res. 53, 431–440 (2009).
Dornetshuber-Fleiss, R. et al. The naturally born fusariotoxin enniatin B and sorafenib exert synergistic activity against cervical cancer in vitro and in vivo. Biochem. Pharmacol. 93, 318–331 (2015).
Liu, S. et al. N-Methylsansalvamide A peptide analogues. Potent new antitumor agents. J. Med. Chem. 48, 3630–3638 (2005).
Otrubova, K. et al. Synthesis and novel structure-activity relationships of potent sansalvamide A derivatives. Chem. Commun. 1033–1034 (2006).
Ujiki, M. B. et al. A novel peptide sansalvamide analogue inhibits pancreatic cancer cell growth through G0/G1 cell cycle arrest. Biochem. Biophys. Res. Commun. 340, 1224–1228 (2005).
Liu, S. et al. Effect of the 4′-substituted phenylalanine moiety of sansalvamide A peptide on antitumor activity. Med. Chem. Commun. 5, 463–467 (2014).
Strobel, G. A., Hess, W. M., Ford, E., Sidhu, R. S. & Yang, X. Taxol from fungal endophytes and the issue of biodiversity. J. Ind. Microbiol. 17, 417–423 (1996).
Shweta, S. et al. Endophytic fungal strains of Fusarium solani, from Apodytes dimidiata E. Mey. ex Arn (Icacinaceae) produce camptothecin, 10-hydroxycamtothecin and 9-methoxycamtothecin. Phytochemistry 71, 117–122 (2010).
Shrivastava, A. et al. Biotechnological advancement in isolation of anti-neoplastic compounds from natural origin: a novel source of L-asparaginase. Acta Biomed. 81, 104–108 (2010).
Kour, A. et al. Isolation and identification of an endophytic strain of Fusarium oxysporum producing podophyllotoxin from Juniperus recurva. World J. Microbiol. Biotechnol. 24, 1115–1121 (2008).
Pérez Hemphill, C. F., Daletos, G., Liu, Z., Lin, W. H. & Proksch, P. Polyketides from the Mangrove-derived fungal endophyte Pestalotiopsis clavispora. Tetrahedron Lett. 57, 2078–2083 (2016).
Fuchser, J. & Zeeck, A. Aspinolides and aspinonene/aspyrone co-metabolites, new pentaketides produced by Aspergillus ochraceus. Liebigs Ann. 1997, 87–95 (1997).
Bode, H. B., Bethe, B., Höfs, R. & Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. Chembiochem. 3, 619–627 (2002).
Gao, S. S. et al. Secondary metabolites produced by solid fermentation of the marine-derived fungus Penicillium commune QSD-17. Biosci. Biotechnol. Biochem. 76, 358–360 (2012).
Wang, J., Debbab, A., Pérez Hemphill, C. F. & Proksch, P. Optimization of enniatin production by solid-phase fermentation of Fusarium tricinctum. Z. Naturforsch. C 68c, 223–230 (2013).
Chávez, R., Fierro, F., García-Rico, R. O. & Vaca, I. Filamentous fungi from extreme environments as a promising source of novel bioactive secondary metabolites. Front. Microbiol. 6, 903 (2015).
Hammerschmidt, L. et al. Cytotoxic acyl amides from the soil fungus Gymnascella dankaliensis. Bioorg. Med. Chem. 23, 712–718 (2015).
Wang, H. et al. Targeted solid phase fermentation of the soil dwelling fungus Gymnascella dankaliensis yields new brominated tyrosine-derived alkaloids. RSC Adv. 85, 81685–81693 (2016).
Nenkep, V. et al. Induced production of bromomethylchlamydosporols A and B from the marine-derived fungus Fusarium tricinctum. J. Nat. Prod. 73, 2061–2063 (2010).
Sørensen, J. L., Akk, E., Thrane, U., Giese, H. & Sondergaard, T. E. Production of fusarielins by Fusarium. Int. J. Food Microbiol. 160, 206–211 (2013).
Kobayashi, H., Sunaga, R., Furihata, K., Morisaki, N. & Iwasaki, S. Isolation and structures of an antifungal antibiotic, fusarielin A, and related compounds produced by a Fusarium sp. J. Antibiot. 48, 42–52 (1995).
Someno, T. et al. ICM0301s, New angiogenesis inhibitors from Aspergillus sp. F-1491. II Physico-chemical properties and structure elucidation. J. Antibiot. 57, 104–109 (2004).
Gai, Y., Zhao, L. L., Hu, C. Q. & Zhang, H. P. Fusarielin E, a new antifungal antibiotic from Fusarium sp. Chin. Chem. Lett. 18, 954–956 (2007).
Sørensen, J. L. et al. Production of novel fusarielins by ectopic activation of the polyketide synthase 9 cluster in Fusarium graminearum. Environ. Microbiol. 14, 1159–1170 (2012).
Chen, H. et al. A new fusarielin analogue from Penicillium sp. isolated from the Mediterranean sponge Ircinia oros. Tetrahedron Lett. 56, 5317–5320 (2015).
Nguyen, H. P. et al. Dehydroxychlorofusarielin B, an antibacterial polyoxygenated decalin derivative from the marine-derived fungus Aspergillus sp. J. Nat. Prod. 70, 1188–1190 (2007).
Fujimoto, H., Aoyama, H., Noguchi-Yachide, T., Hashimoto, Y. & Kobayashi, H. Fusarielin A as an Anti-angiogenic and anti-proliferative agent: basic biological characterization. Chem. Pharm. Bull 56, 298–304 (2008).
Ola, A. R. B., Thomy, D., Lai, D., Brötz-Oesterhelt, H. & Proksch, P. Inducing secondary metabolite production by the endophytic fungus Fusarium tricinctum through coculture with Bacillus subtilis. J. Nat. Prod. 76, 2094–2099 (2013).
Yang, S. X. et al. Toxic lanostane triterpenes from the basidiomycete Ganoderma ambionense. Phytochem. Lett. 5, 576–580 (2012).
Shao, C. L. et al. Potent Antifouling Resorcylic acid lactones from the gorgonian-derived fungus Cochliobolus lunatus. J. Nat. Prod. 74, 629–633 (2011).
Paranagama, P. A., Wijeratne, E. M. K. & Gunatilaka, A. A. L. Uncovering biosynthetic potential of plant-associated fungi: Effect of culture conditions on metabolite production by Paraphaeosphaeria quadriseptata and Chaetomium chiversii. J. Nat. Prod. 70, 1939–1945 (2007).
Zhang, J., Liu, D., Wang, H., Liu, T. & Xin, Z. Fusartricin, a sesquiterpenoid ether produced by an endophytic fungus Fusarium tricinctum Salicorn 19. Eur. Food Res. Technol. 240, 805–814 (2015).
Pérez Hemphill, C. F. et al. Absolute configuration and anti-tumor activity of torrubiellin B. Tetrahedron Lett. 56, 4430–4433 (2015).
Kjer, J., Debbab, A., Aly, A. H. & Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 5, 479–490 (2010).
Gosepath, E. M. et al. Acquired cisplatin resistance in the head-neck cancer cell line Cal27 is associated with decreased DKK1 expression and can partially be reversed by overexpression of DKK1. Int. J. Cancer 123, 2013–2019 (2008).
Engelke, L. H., Hamacher, A., Proksch, P. & Kassack, M. U. Ellagic acid and resveratrol prevent the development of cisplatin resistance in the epithelial ovarian cancer cell line A2780. J. Cancer 7, 353–362 (2016).
Acknowledgements
PP and RSO acknowledge support by the International Scientific Partnership Program ISPP 0065 at King Saud University. PP wants to thank the GRK 2158 supported by the DFG for financial support. MUK would like to thank the Bundesministerium für Bildung und Forschung (BMBF) for financial support (BMBF-16GW0108). We are grateful to the Institute of Pharmaceutics and Biopharmaceutics (HHU, Duesseldorf) for providing the osmometer, and to the Department of Chemistry (HHU, Duesseldorf) for performing NMR and HRESIMS measurements.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Hemphill, C., Sureechatchaiyan, P., Kassack, M. et al. OSMAC approach leads to new fusarielin metabolites from Fusarium tricinctum. J Antibiot 70, 726–732 (2017). https://doi.org/10.1038/ja.2017.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.21
This article is cited by
-
Rubiflavin G, photorubiflavin G, and photorubiflavin E: Novel pluramycin derivatives from Streptomyces sp. W2061 and their anticancer activity against breast cancer cells
The Journal of Antibiotics (2023)
-
Challenges and Opportunities for Bioactive Compound and Antibiotic Discovery in Deep Space
Journal of the Indian Institute of Science (2023)
-
Natural products in drug discovery: advances and opportunities
Nature Reviews Drug Discovery (2021)
-
Fungal endophytes in Peperomia obtusifolia and their potential as inhibitors of chickpea fungal pathogens
World Journal of Microbiology and Biotechnology (2021)
-
Induction of metabolic variability of the endophytic fungus Xylaria sp. by OSMAC approach and experimental design
Archives of Microbiology (2021)
