
Abstract
A novel, practical and stereoselective synthesis of (3R,4R)-4-acetoxy-3-[(R)-1-(t-butyldimethylsilyloxy)ethyl]-2-azetidinone, a key intermediate in the preparation of β-lactam antibiotics is reported. The crucial step of the synthesis is based on the Cu(I)-mediated Kinugasa cycloaddition/rearrangement cascade between silyl protected (R)-3-butyn-2-ol and the nitrone derived from benzyl hydroxylamine and benzyl glyoxylate. The obtained adduct is subjected to debenzylation with sodium, or lithium in liquid ammonia followed by oxidation with lead tetraacetate to afford the final product.
Similar content being viewed by others
Introduction
Penems and carbapenems are important β-lactam antibiotics that continuously provoke interest within academic and pharmaceutical laboratories.1, 2 They show excellent activities against both Gram-positive and Gram-negative bacteria, while being resistant towards hydrolysis by most β-lactamases.3
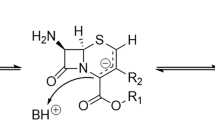
A common key intermediate for their synthesis is 4-acetoxyazetidinone 1 (Scheme 1).4 This molecule contains three stereogenic centers and a AcO group at the C-4 position of the four-membered β-lactam ring. The AcO group can be easily substituted by a variety of nucleophiles.4 These structural features lay the foundation for the possibility of transforming azetidinone 1 into an array of penem and carbapenem antibiotics.4, 5
Azetidinone 1 has been obtained by diverse chiral approaches.4, 5 The earliest references to azetidinone 1 originated from Sankyo Co. Ltd., and employed penicillanate esters as the chiral source.6 However, the most convenient method for the preparation of azetidinone 1 is the asymmetric [2+2] cycloaddition of a diketene with chiral imines derived from various chiral pools, including ethyl (S)-lactate,7 L-menthyl glyoxylate,8 and D-mannitol.9
A relatively short route to azetidinone 1 has been reported by Ohashi of Kanegafuchi Chemical Industries.10, 11, 12 The initial (R)-3-hydroxybutyrate was transformed into terminal trimethylsilyl-vinyl ether that subsequently was subjected to [2+2] cycloaddition with chlorosulfonyl isocyanate to afford the target azetidinone 1.
Another interesting approach was described by Tatsuta et al.13 detailing the synthesis of azetidinone 1 from methyl 6-deoxy-glucosamine.
Results and discussion
Recently, we have reported direct and simple approach to the formation of the basic skeleton of carbapenams14, 15, 16, 17 and N,4-diaryl substituted β-lactam18 frameworks via diastereoselective copper(I)-mediated reaction between nitrones and terminal acetylenes. In most cases, reactions displayed high diastereoselectivity leading to one dominant product.19
Herein, we describe a short, high yielding and stereoselective synthesis of azetidinone 1 in which the pivotal step, the formation of 2-azetidinone ring, is Cu(I)-mediated Kinugasa cycloaddition/rearrangement cascade between terminal acetylene 2 and nitrone, 3 which is derived from benzyl glyoxalate and N-benzyl hydroxylamine (Scheme 2).
The reaction proceeds in acetonitrile under our standard conditions to provide azetidinone 4 in 73% yields as a mixture of two diastereomers cis -4 and trans- 4 in a ratio of ∼3:1, respectively. The proportion of cis/trans diastereomers of azetidinone 4 was determined by 1H NMR. Nitrone 3 exists in solution as an easily convertible mixture of Z and E isomers in a ratio, which is solvent dependent.20 One can assume that the first step in Kinugasa cascade, 1,3-dipolar cycloaddition, proceeds preferentially with more reactive rotamer. Our earlier observations of Kinugasa reaction between acetylene 2 and non-chiral nitrones suggested that β-lactam (S)-configuration should be obtained at the C-3 carbon atom.14 Formation of a relatively large amount of trans isomer (trans- 4) could be due to base catalyzed epimerization of the initially formed cis- 4, which can occur under these reaction conditions. Consequently, cis (4R) and trans (4R) substituted β-lactams are assigned. Correct configurational assignments were proved at the end of the synthesis by comparison with target product with known, commercially available samples of azetidinone 1.
The isomeric mixture 4 was subjected to sodium or lithium reduction in liquid ammonia to provide acid 5 in 98% yield, as a mixture of diastereomers (Scheme 3). Acid 5 was subsequently oxidized by treatment with Pb(OAc)4 in AcOH21 to provide the desired azetidinone 1 in 73% yield, with only minute amounts of the by-product 6 observed. Oxidation of acid 5 proceeds with transformation of both cis and trans isomers into acetate 1 possessing a trans substituted ring. Azetidinone 1 displays the same analytical and spectral data as commercially available (3R,4R)-4-acetoxy-3-[(R)-1-(t-butyldimethylsilyloxy)ethyl]-2-azetidinone (Aldrich, Cat. no. 375845 [CAS 76855-69-1]).
Bearing in mind readily available starting materials 2 and 3, as well as three simple steps toward azetidinone 1, the reported synthesis offers some advantages over many so far reported.
Experimental Procedure
1H and 13C NMR spectra were recorded on Varian VN MRS Spectrometer (Varian Inc., St. Clara, CA, USA) at 600 and 150 MHz, respectively, in CDCl3 or C6D6 with tetramethylsilane (TMS) as an internal standard. Chemical shifts are reported as δ values in p.p.m., and coupling constants are in hertz. IR spectra were recorded on a FT-IR-1600 Perkin-Elmer spectrophotometer (Perkin Elmer, Waltham, MA, USA). Optical rotation was recorded on Jasco P-2000 polarimeter (Jasco Inc., Easton, MD, USA). High-resolution mass spectra were recorded on Synapt G2-S Waters Spectrometer for ESI. (R)-(+)-3-Butyn-2-ol and dibenzyl L-tartrate were purchased from TCI Europe (Zwijndrecht, Belgium).

Azetidinone 1 as a building block for synthesis of beta-lactam antibiotics.

Retrosynthetic analysis.

Synthesis of azetidinone 1.
Acetylene 2
Tert-butyldimethylchlorosilane (TBSCl) (0.828 g, 5.5 mmol) was added portionwise to a solution of (R)-(+)-3-butyn-2-ol (0.350 g, 5.0 mmol) and imidazole (0.374 g, 5.5 mmol) in dry CH2Cl2. After 2 h, the mixture was diluted with CH2Cl2 (10 ml) and washed with water (5 ml). The organic phase was dried over anhydr. Na2SO4 and the solvent was removed under slightly diminished pressure (383 hPa). The residue was dissolved in pentanes and filtered through a short silica gel pad to afford acetylene 2 (0.828 g, 90%). [α]D +46.2 (c 1, CH2Cl2) 1H NMR (600 MHz, CDCl3) δ: 4.51 (1H, dq, J 6.4, 2.0 Hz), 2.57 (1H, d, J 2.0 Hz), 1.41 (3H, d, J 6.4 Hz, CH3), 0.9 (s, 9H, tBu), 0.13 (s, 3H, SiCH3), 0.12 (s, 3H, SiCH3).
Benzyl gloxalate
A 500-ml, one-necked round-bottomed flask (equipped with nitrogen inlet) was charged with dibenzyl L-tartrate (17.3 g, 52.4 mmol) and 300 ml of anhydrous ethyl ether. To the resulting solution periodic acid dihydrate was added (11.9 g, 52.4 mmol). The slurry was stirred at 23 °C for 1.5 h during which a white precipitate had formed. The solution was filtered through Celite (washing with 70 ml diethyl ether) and the filtrate was concentrated in vacuo (not to dryness) to give benzyl glyoxylate, which was used without any further purification. 1H NMR (600 MHz, C6D6) δ: 8.86 (s, 1H), 7.27–7.45 (m, 5H), 5.10 (s, 2H).
Nitrone 3
Benzyl glyoxalate (7.34 g, 56.9 mmol) was dissolved in MeOH (60 ml) and anhydr. AcONa (6.07 g, 74.0 mmol) was added at room temperature. Next, a solution of N-benzylhydroxylamine hydrochloride (9.08 g, 56.9 mmol) in MeOH (20 ml) was added dropwise. After 2 h, MeOH was removed and the residue was partitioned between CH2Cl2 (100 ml) and water (30 ml). The organic phase was dried over MgSO4 and then the solvent was removed to give 3 (14.5 g) as a white solid. The solid was recrystallized from hexanes/CH2Cl2 mixture to provide 12 g (yield 78%) of nitrone 3 as colorless crystals. M.p. 105–106 °C; 1H NMR (600 MHz, CD3CN) δ: 7.30–7.15 (m, 10H), 5.18 (s, 2H), 5.00 (s, 2H) 2.15 (s, 1H).
Azetidinone 4
Triethylamine (0.56 ml, 4.0 mmol) was added to a suspension of CuI (190 mg, 1.0 mmol) in degassed MeCN (4 ml). Subsequently, the mixture was cooled to 0 °C and acetylene 2 (184 mg, 1.0 mmol) in MeCN (1 ml) was added. After 15 min, a solution of nitrone 3 (404 mg, 1.5 mmol) in MeCN (5 ml) was added slowly. The mixture was kept at 0 °C for an additional 15 min. After removal of the cooling bath, the mixture was stirred at ambient temperature under an inert atmosphere for 24 h. The progress of the reaction was monitored by TLC. Removal of the solvent gave a residue, which was chromatographed on silica gel (hexane/AcOEt 4:1 v/v) to afford a mixture of azetidinones cis-4 and trans-4 in ratio 3:1 (323 mg, 71%, according to 1H NMR of crude reaction mixture). cis-4: 1H NMR (600 MHz, C6D6) δ: 7.10–6.94 (10H, m, Ar), 5.06 (1H, ABq, J 12.2 Hz, CH2 Ph), 4.93 (1H, ABq, J 12.2 Hz CH2 Ph), 4.76 (1H, ABq, J 15.4 Hz, NCH2Ph), 4.29 (1H, ddd, J 3.6, 6.6, 13.0 Hz, H-5), 4.15 (1H, ABq, J 15.4 Hz, NCH2Ph), 3.59 (1H, d, J 5.8 Hz, H-4), 2.81 (1H, dd, J 3.6, 5.8 Hz, H-3), 1.10 (3H, d, J 6.6 Hz, CH3), 0.96 (9H, s, t-Bu), 0.48 (3H, s, SiCH3), 0.16 (3H, s SiCH3); 13C NMR (150 MHz, C6D6) δ: 169.1, 165.9, 135.7, 135.5, 66.4, 65.0, 64.5, 62.0, 52.6, 44.6, 25.73, 25.66, 21.6, 20.7, 18.0, −0.4, −4.6, −5.04; IR (CH2Cl2) v: 2929, 1766, 1257, 1197, 837 cm−1. trans-4: 1H NMR (600 MHz, C6D6 ) δ: 4.86 (1H, ABq, J 12.2 Hz, CH2Ph), 4.74 (1H, ABq, J 12.2 Hz CH2 Ph), 4.61 (1H, ABq, J 14.9 Hz NCH2Ph), 4.12 (1H, ABq, J 14.9 Hz NCH2Ph), 4.12 (1H, d, J 2.6, H-4), 4.00 (1H, ddd, J 3.6, 6.3, 12.6 Hz, H-5), 3.01 (1H, dt, J 0.7, 3.3 Hz, H-3), 0.94 (3H, d, J 6.2 Hz, CH3), 0.84 (9H, s, t-Bu), 0.16 (3H, s, SiCH3), −0.04 (3H, s, SiCH3); 13C NMR (150 MHz, C6D6) δ: 170.5, 165.4, 135.5, 135.4, 65.4, 64.5, 63.6, 62.5, 52.0, 45.2, 25.6, 22.1, 20.7, 17.8, −4.9, −4.9; HR MS (ESI) m/z calcd for [M+ Na]+ C26H35NO4NaSi: 476.2228; Found: 476.2236; anal calcd for C26H35NO4Si (%): C, 68.84, H, 7.78, N, 3.09; Found C, 68.87, H, 7.79, N, 3.19.
Azetidinone 5
A solution of azetidinone 4 (150 mg, 0.33 mmol) in tetrahydrofurane (THF) (2 ml) was added to a cooled to −78 °C mixture of lithium (36 mg, 4.06 mmol) in dry liquid NH3. After 25 min, the reaction was quenched by addition of solid NH4Cl (0.825 g). The reaction mixture was gradually warmed to ambient temperature. Next, CH2Cl2 (41 ml) was added and the resulting solid was filtered off. After removal of solvent, the residue was dissolved in Et2O-water mixture (1:1). The aqueous solution was lyophilized to afford a mixture of cis-5 and trans-5 (88 mg, 98%, ratio 3:1) as a white solid. cis-5: 1H NMR (600 MHz, D2O) δ: 4.16 (1H, ddd, J 5.1, 11.3 Hz, H-5), 4.05 (1H, d, J 5.8 Hz, H-4), 3.49 (1H, dd, J 5.8, 5.2 Hz, H-3), 1.14 (3H, d, J 6.4 Hz, CH3), 0.74 (9H, s, Sit-Bu), 0.01 (3H, s, SiCH3), 0.02 (3H, s, SiCH3); 13C NMR (150 MHz, D2O) δ: 176.1, 171.8, 65.5, 60.1, 53.0, 25.4, 25.1, 21.3, 20.4, 17.5, –5.05, –5.55. trans-5: 1H NMR (600 MHz, D2O) δ: 4.22 (1H, ddd, J 2.8, 6.4, 12.8 Hz, H-5), 4.02 (1H, d, J 2.3 Hz, H-4), 3.06 (1H, t, J 2.6 Hz, H-3), 1.15 (3H, d, J 6.4 Hz, CH3), 0.74 (9H, s, Sit-Bu), 0.01 (3H, s, CH3), 0.02 (3H, s, CH3), MS (ESI): [M+Na]+ 296.1; MS (ESI): [M−H]− 272.2, [M+HCOO]− 318.1.
Azetidinone 1
To a solution of azetidinones 5 (90 mg, 0.331 mmol) in dry N,N-dimethylformamide (DMF) (1.7 ml), AcOH (322 μl) and Pb(OAc)4 (219 mg, 0.494 mmol) was added. After 3 h, DMF and AcOH were removed under diminished pressure and the residue was treated with Et2O (15 ml). The remaining solids were removed and the solvent was evaporated to provide azetidinone 1 (15 mg, 73%). Mp. 105–107 °C [Lit. 107–109 °C, (Aldrich, Cat. no. 375845 [CAS 76855-69-1])]; [α]D +51.7 (c 0.4, CHCl3) [Lit. [α]D +51 (c 1, CHCl3), (Aldrich, Cat. no. 375845 [CAS 76855-69-1])]; 1H NMR (600 MHz, C6D6) δ: 5.74 (1H, d, J 1.0 Hz, H-4), 5.70 (1H, bs, NH), 3.89 (1H, ddd, J 3.8, 6.3, 12.6 Hz, H-5), 2.87 (1H, dd, J 1.0, 3.8 Hz, H-3), 1.50 (3H, s, OAc), 0.99 (3H, d, J 6.3 Hz, CH3), 0.90 (9H, s, t-BuSi), 0.06 (3H, s, CH3Si), −0.04 (3H, s, CH3Si); 13C NMR (150 MHz, C6D6) δ: 170.1, 165.7, 75.1, 65.3, 64.1, 25.6, 21.9, 19.9, 17.8, −4.7, −5.3. HR MS (ESI) m/z calcd for C13H25NO4SiNa [M+Na]+ 310.1451; found 310.1459.
References
Morin, R. B. & Gorman, M. Chemistry and Biology of β-Lactam Antibiotics, Academic Press: New York, NY, USA, (1982).
Georg, G. I. Organic Chemistry of β-lactams, VCH, Weinheim, (1993).
Banik, B. K. (ed.) in Top. Heterocycl. Chem., Springer, (2010).
Berks, A. H. Preparations of two pivotal intermediates for the synthesis of 1-β-methyl carbapenem antibiotics. Tetrahedron 52, 331–375 (1996).
Hungerbuhler, E. et al in New Aspects of Organic Chemistry I. New Approaches to Versatile Chiral β-Lactam Building Blocks; Application in the Synthesis of Novel Antibiotics, VCH Publishers: Tokyo, (1989) (and references cited therein).
Yoshida, A., Hayashi, T., Takeda, N., Oida, S. & Ohki, E. 2-(Alkylthio)penem-3-carboxylic acids. IV. Synthesis of (hydroxyethyl)-azetidinone precursors to 1-thia analogs of thienamycin. Chem. Pharm. Bull. 29, 2899–2909 (1981).
Ito, Y., Kawabata, T. & Terashima, S. A novel synthesis of (3R,4R)-4-acetoxy-3-[(R)-1-(t-butyldimethylsilyloxy)ethyl]-2-azetidinone, the versatile key intermediate of carbapenem synthesis, from (S)-ethyl lactate. Tetrahedron Lett. 27, 5751–5754 (1986).
Sasaki, A., Goda, K., Enomoto, M. & Sunagawa, M. Synthetic studies of carbapenem and penem antibiotics. II. Synthesis of 3-acetyl-2-azetidinones by [2+2]-cycloaddition of diketene and Schiff bases. Chem. Pharm. Bull. 40, 1094–1097 (1992).
Yang, M. G., Jung, H. L., Jin, S. C. & Young, Y. L. Synthesis of 3-acetyl-2-azetidinones by [2+2] cycloaddition of diketene and imines. Bull. Korean Chem. Soc. 17, 985–987 (1996).
Ohashi, T. et al. U.S. Patent US 4861877 (1989).
Ohashi, T. et al. U.S. Patent US 4791198 (1988).
Ohashi, T. et al. U.S. Patent US 5061817 (1991).
Tatsuta, K., Takahashi, M., Tanaka, N. & Chikauchi, K. Novel synthesis of (+)-4-acetoxy-3-hydroxyethyl-2-azetidinone from carbohydrate. A formal total synthesis of (+)-thienamycin. J. Antibiot. 53, 1231–1234 (2000).
Michalak, M. et al. A formal synthesis of ezetimibe via Kinugasa cycloaddition/rearrangement cascade. J. Org. Chem. 76, 6931–6936 (2011).
Mames, A. et al. Direct, catalytic synthesis of carbapenams via cycloaddition/rearrangement cascade reaction: unexpected acetylenes’ structure effect. J. Org. Chem. 75, 7580–7587 (2010).
Stecko, S., Mames, A., Furman, B. & Chmielewski, M. Asymmetric Kinugasa reaction of cyclic nitrones and non-racemic acetylenes. J. Org. Chem. 74, 3094–3100 (2009).
Stecko, S., Mames, A., Furman, B. & Chmielewski, M. Diastereoselective synthesis of carbapenams via Kinugasa reaction. J. Org. Chem. 73, 7402–7404 (2008).
Michalak, M. et al. Synthesis of N,4-diaryl substituted β-lactams via Kinugasa cycloaddition/rearrangement reaction. Tetrahedron 68, 10806–10817 (2012).
Maciejko, M. et al. An entry to carbapenem antibiotics scaffold via asymmetric Kinugasa reaction. Synthesis 2825–2839 (2012).
Stanko, J. A. The design and synthesis of novel chiral Z-nitrones with applications towards the syntheses of enantiomerically pure 4-hydroxy amino acids PhD thesis, Duke Univ. (2009).
Zhou, G. B., Guan, Y. Q., Tang, H., Zhao, Y. B. & Yang, L. R. Practical synthetic approach to 4-acetoxy-2-azetidinone for the preparation of carbapenem and penem antibiotics. Res. Chem. Intermed. 38, 251–259 (2012).
Acknowledgements
The authors are grateful to the European Union within European Regional Development Fund (Project POIG.01.01.02.-14-102/09) for financial support of research.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is dedicated to Professor Kuniaki Tatsuta commemorating his 101 total synthesis.
Rights and permissions
About this article
Cite this article
Grzeszczyk, B., Stecko, S., Mucha, Ł. et al. A practical preparation of the key intermediate for penems and carbapenems synthesis. J Antibiot 66, 161–163 (2013). https://doi.org/10.1038/ja.2013.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2013.8
