
Abstract
Three new angucyclinones, saccharosporones A, B and C, together with (+)-ochromycinone, (+)-rubiginone B2, tetrangulol methyl ether and fujianmycin A, were obtained from fermentation of the terrestrial actinomycete of the genus Saccharopolyspora BCC 21906 isolated from a soil collected in Chanthaburi Province, Thailand. Structures of the new compounds and their relative configurations were assigned by NMR spectral data interpretation. Saccharosporones A and B exhibited antimalarial activity against Plasmodium falciparum K1 with IC50 values of 4.1 and 3.9 μM. Both metabolites also possessed cytotoxic activities against cancer cell lines (KB, MCF-7 and NCI-H187) and nonmalignant Vero cell, while saccharosporone C only showed cytotoxic activity against NCI-H187.
Similar content being viewed by others


Introduction
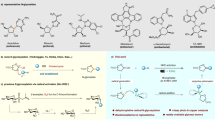
As angucycline metabolites are a large class of antibiotics with a broad spectrum of biological activities and interesting chemical structures, they have attracted much attention.1, 2 In general, a compound from this family is characterized by an angular tetracyclic framework that possesses a benz[a]anthracene ring system. In the course of our screening program of rare actinomycetes in Thailand for novel bioactive secondary metabolites, fermentation of Saccharopolyspora sp. BIOTEC Culture Collection (BCC) 21906 led to the isolation of three new angucyclines saccharosporones A (1), B (2) and C (3), (Figure 1) and several known metabolites, including ochromycinone (4),3, 4 (+)-rubiginone B2 (5),5 tetrangulol methyl ether (6)6, 7 and fujianmycin A.8 In this paper, we report the isolation, structure elucidation and biological activities of these new angucyclinone polyketides.

The structures of saccharosporones A (1), B (2) and C (3) from Saccharopolyspora sp. BCC 21906.
The culture of Saccharopolyspora sp. BCC 21906 was extracted exhaustively with ethyl acetate. The crude substance was evaporated to dryness and fractionated by partition on the HP-20SS resin, subsequently eluted with acetone/water mixtures to obtain 20, 30, 40, 50, 60, 70, 80, 90 and 100% acetone fractions. Further purification of 70% and 80% acetone–water fractions by reversed-phase C18 HPLC furnished compounds 1–3 as minor constituents.
Results and discussion
Saccharosporone A (1) was obtained as a yellowish powder and its molecular formula was established as C19H20O5 by high-resolution HRESITOFMS (m/z 327.1231, [M −H]−) and its NMR data. The LRESIMS spectra showed ions corresponding to [M+Na]+ and [M+H]+ at m/z 351 and 329, respectively. Analysis of the IR spectroscopic data showed an absorption band at 3474 cm−1, indicating hydroxyl functionality, and a signal at 1632 cm−1, which suggested the presence of a chelated quinone carbonyl group.9 The violet color reaction with the addition of sodium hydroxide indicated a peri-hydroxy quinone functional group.
The 1H NMR spectrum of 1, measured in acetone-d6 (Table 1), illustrated three olefinic/aromatic proton signals at δ 7.26, 7.58 and 7.71, three hydroxy proton signals at δ 3.65, 4.24 and 12.14, in addition to one methyl, three methine and four methylene proton signals. The analysis of 1H–1H COSY NMR spectrum showed three coupled spin systems. The first spin system consisted of three doublets of doublets at δ 7.26 7.58 and 7.71, which can be attributed to 1,2,3-trisubstituted aromatic ring system. The second spin system involved a hydroxy (δ 4.24)-bearing methine (δ 4.04) adjacent to a methylene group (δ 2.88–2.90). The final spin system had a methine (δ 2.77) connected to two correlated methylenes (δ 1.06, 1.70 and 1.92) attached to a methine at δ 2.29 that also coupled to a methyl group (δ 0.89) and another methylene (δ 1.38 and 2.06).
The 13C NMR/DEPT spectrum of 1 showed 19 carbon resonances of a methyl group (δ 22.4), 2 carbonyl carbons (δ 183.5 and 190.8), 3 non-oxygenated olefinic/aromatic methine carbons (δ 118.4, 123.2 and 136.2), 5 olefinic/aromatic quaternary carbons, including 1 bearing oxygen, 1 oxygenated quaternary carbon, 3 methines and 4 methylene carbons. The major portion of the angucyclinone backbone could be assembled through the interpretation of COSY and HMBC correlations (Figure 2). The cross-peaks between H-9 (δ 7.26) and H-10 (δ 7.71) to C-8 (δ 161.1) suggested the presence of an oxygenated carbon in the aromatic ring. The 1H NMR signal at δ 12.14 could be assigned to a chelated peri-hydroxy group attached to C-8 according to its HMBC correlations to the carbons at C-7a (δ 114.9), C-8 and C-9 (δ 123.2). The aromatic methine at H-11 (δ 7.58) correlated to carbonyl at C-12 (δ 183.5) indicated the carbonyl position adjacent to the trisubstituted aromatic ring. Both methine signal at H-12b (δ 2.77) and methylene protons at H2-6 (δ 2.88–2.90) showed HMBC correlations to the same carbons at C-4a (δ 69.1), C-5 (δ 72.7), C-6a (δ 141.1) and C-12a (δ 147.4), while H-12b correlated to a carbonyl carbon at C-12 and H-6 to a carbonyl at C-7 (δ 190.8), confirming the second spin system next to the quinone ring. The HMBC correlations from methine proton at H-5 (δ 4.04) to carbons at C-4a, C-6a and C-12b (δ 42.3) and COSY correlations between δ 4.04 and δ 4.24 established a secondary alcohol at C-5, whereas HMBC correlations from hydroxyl proton at δ 3.65 to C-5 and C-12b supported the position of a tertiary alcohol at C-4a. Analysis of the COSY allowed the assignment of C-12b/C-1/C-2/C-3/C-4 connectivities, which made up a major portion of the third spin system. In addition, methylene signals at H2-4 (δ 1.38, 2.06) showed correlations to carbons at C-4a, C-5 and C-12b, while the other methylene at δ 1.92 correlated to C-4a, C-12a and C-12b, establishing the core structure of the third spin system. Unlike the other angucyclinone polyketides, saccharosporone A lacked a carbonyl group at C-1 but instead contained a secondary alcohol at C-5 position.

Selected COSY (bold lines) and HMBC (arrows) correlations observed for 1, 2 and 3.
An attempt to determine the absolute configuration of 1 was carried out using Mosher’s method under several conditions but none of the derivatization reactions was successful, possibly because of the steric hindrance of secondary alcohol in the axial orientation. The relative stereochemistry of molecule 1 was determined by the interpretation of the NOESY NMR spectroscopic data. The strong NOESY correlations from methylene signal at He-4 (δ 2.06) to methine protons at H-3 (δ 2.29) and H-5 (δ 4.04) suggested that these protons were on the same side of the ring. In addition, the important correlations between a methine proton H-12b (δ 2.77) and two methylene signals at Ha-2 (δ 1.06) and Ha-4 (δ 1.38), along with the fact that no correlation between H-5 and H-12b has been observed, confirmed that H-5 and H-12b were on the opposite side of the ring. The cis relative configuration of two hydroxyl groups at C-4a and C-5 could be proposed on the basis of the analysis by NOESY and 1D NOE difference spectrum. Irradiation of the methylene proton at H2-1 (δ 1.92) showed correlation to H2-6 (δ 2.88–2.90), which supported this arrangement (Figure 3).

Assigned relative configuration of 1 from selected NOESY NMR correlations.
Saccharosporone B (2) was obtained as a yellowish powder and its structure was also elucidated on the basis of the interpretation of 2D NMR data. In comparison to 1, both 1H and 13C NMR chemical shifts were quite comparable (Table 1), except the major difference at C-1 (δ 83.5) attributed to oxygenated methine (δ 4.25) instead of a methylene group. The correlation patterns of the COSY and HMBC NMR spectral data were revealed to be very similar to 1 (Figure 2). The molecular formula of 2 was established as C19H20O6 by HRESITOFMS and only differed from 1 by having one additional hydroxy group instead. This conclusion was supported by its 13C NMR chemical shift in the downfield region at C-1. Therefore, saccharosporone B (2) could be established as a 1-hydroxy analogue of 1. The relative configuration of 2 was also derived based on the interpretation of NOESY spectrum, and the same correlation pattern has been observed in comparison to 1. The methine proton at H-12b (δ 3.04) showed correlations to the methylene signals at Ha-2 (δ 1.17) and Ha-4 (δ 1.64), while the oxygenated methine at H-5 (δ 4.09) correlated to the methylene proton He-4 (δ 2.09–2.11) and a methine signal at H-3 (δ 2.12–2.14). Accordingly, the configuration of two hydroxy groups at C-4a (δ 73.7) and C-5 (δ 76.7) could be proposed to stay on the same ring side as similar to 1. The presence of a cross-peak between H-1 (δ 4.25) and H-12b, whereas no correlation between H-1 and Ha-3 has been recorded, confirming that the additional secondary alcohol at C-1 was on the opposite side of the ring when compared with the other two hydroxy groups at C-4a and C-5. Both 1 and 2 possessed similar relative configurations as well as their optical rotations that exhibited the same negative sign. The color reaction with sodium hydroxide from yellow to violet also supported the presence of a peri-hydroxy quinone functional group.
Compound 3, saccharosporone C, was also obtained as a yellowish powder and its structure was found to be closely related to compound 1. The 1H and 13C NMR spectra of 3 is mostly similar to 1 with the same 1,2,3-trisubstituted aromatic ring. The major difference is the change in the quinone ring, as only one carbonyl carbon remained at C-12 (δ 200.3), whereas C-7 (δ 62.8) has been altered to an oxygenated methine signal. Furthermore, two olefinic quaternary carbons, which formed the double bond between C-6a and C-12a, have been replaced with two aliphatic methine carbons. A molecular formula of C19H24O5 of 3 was established by HRESITOFMS (m/z 333.1697, [M+H]+). Analysis of the COSY data allowed the assignment of the C-12a/C-6a/C-7/hydroxyl group (δ 3.99) connectivities, and HMBC correlations from H-7 (δ 5.07) to C-11a (δ 133.7) and C-12a (δ 43.1) further confirmed that a carbonyl at C-7 in 1 and 2 has been replaced by a methine carbon attached to a hydroxy group, while the double bond between C-6a and C-12a became saturated. The relative configuration of 3 was proposed on the basis of NOESY spectrum and by analysis of vicinal coupling constants. The configurations at C-3, C-4a and C-12b were established to be similar to those of compound 1. Important correlation between H-6a (δ 2.44) and Ha-1 (δ 1.81) confirmed the assignment of configurations at C-4a and C-12b. The position of a hydroxyl group at C-5 was opposite to 1 and 2 as suggested by the lack of NOESY correlations from H-5 to H-3 and H-6a. The trans configuration at C-6a and C-12a was supported by NOESY correlations from a methine signal at H-6a to Ha-1, He-6 (δ 1.63) and H-7, along with correlations from H-12a (δ 3.37) to Ha-6 (δ 2.60) and H-12b (δ 2.30). Furthermore, the large coupling constant between H-6a and H-12a (JH-6a/H-12a=12.5 Hz) represented a diaxial relationship of these protons. The relative configuration of the hydroxy at C-7 was confirmed by NOESY correlations from H-7 to He-6 (δ 1.63) and H-6a and from the lack of correlation between H-7 and H-12a or Ha-6.
Isolated pure metabolites were screened for antimalarial, antibacterial, antifungal and cytotoxic activities. Compounds 1, 2, 5 and 6 exhibited antimalarial activities against Plasmodium falciparum K1 with respective IC50 values of 4.1, 3.9, 6.0 and 4.6 μM, whereas 1, 2, 4 and 6 showed growth inhibition against Mycobacterium tuberculosis with IC50 values of 76.2, 72.7, 40.8 and 19.7 μM, respectively. None of them exhibited activities against Bacillus cereus, while only 2 displayed antifungal activity against Candida albicans with an IC50 value of 46.2 μM. New angucyclinones 1 and 2 showed cytotoxic activities against all three cancer cell lines (KB, MCF-7 and NCI-H187) while 3 only exhibited weak cytotoxicity against NCI-H187 (Table 2).
Experimental procedures
General
Optical rotations were measured on a JASCO P-1030 digital polarimeter. UV spectra were obtained on a SPEKOL 1200 spectrophotometer (Analytikjena, Jena, Germany). FT-IR spectra were taken on a Bruker Alpha-E spectrometer (Bruker Optik, GmbH, Ettlingen, Germany). 1H, 13C, DEPT, COSY, HMQC, HMBC and NOESY NMR spectra were recorded on a Bruker AV500D spectrometer (Bruker BioSpin AG Fällanden, Switzerland). ESI-TOF mass spectrometer measurements were obtained on a Micromass LCT (Micromass UK Limited, Manchester, UK) and a Bruker micrOTOF mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany).
Biological material
The actinomycete Saccharopolyspora sp. (BCC 21906) was isolated from a soil collected at Evergreen forest, Khao Kitchakut National Park, Chanthaburi Province, Thailand. Analysis of the nucleotide sequence of the 16S rRNA gene of Saccharopolyspora sp. BCC 21906 exhibited a sequence similarity to the genes of S. antimicrobica I05-00074(T) (98.62%) and S. hirsuta subsp. kobensis JCM 9109(T) (98.14%). The collection and taxonomic identification were performed by Mr Chanwit Suriyachadkun (BIOTEC). A specimen has been deposited at BIOTEC Culture Collection and designated as BCC 21906.
Culture conditions
Saccharopolyspora sp. BCC 21906 was maintained on the International Streptomyces Project medium 2 at 28 °C for 20–30 days. Later, the agar was cut into pieces (1 × 1 cm2) and inoculated into 3 × 250 ml Erlenmeyer flasks each containing 25 ml of Bio 19.1 medium (glucose 20 g, peptone 5 g, yeast extract 2 g, meat extract 5 g, NaCl 0.5 g, CaCO3 3 g and vitamin B complex solution 1 ml, per 1 liter of distilled water). After the incubation on a rotary shaker (250 r.p.m.) at 28 °C for 7 days, each primary culture was transferred into a 1000-ml Erlenmeyer flask containing 250 ml of the same liquid medium and incubated at 28 °C for 7 days on a rotary shaker (250 r.p.m.). Each 25 ml portion of the secondary cultures was transferred into 20 × 1 liter Erlenmeyer flasks each containing 225 ml of production medium LS2 (mannitol 20 g, soy meal 20 g and trace element mix 0.25 ml, per 1 liter of distilled water). The mixture of trace elements consisted of (w/v): CaCl2, 0.4%; ZnSO4, 0.2%; Na2B4O7, 0.01%; FeSO4, 0.5%; KI, 0.005%; CoCl2, 0.05%; CuSO4, 0.02%; MnCl2, 0.2%; Na2MoO4, 0.005%; (v/v) H2SO4 (95–97% p.a.), 0.1%. A total of 5 l (20 × 250 ml) of BCC 21906 culture was incubated on rotary shakers (250 r.p.m.) for 7 days at 28 °C.
Extraction and isolation
The organic constituents from a 5 l culture of Saccharopolyspora sp. strain BCC 21906 were extracted with ethyl acetate. The crude organic layer was concentrated in vacuo to yield a dark substance (3.7 g), which was partitioned by Diaion HP20SS (Supelco Bellefonte, PA, USA) column chromatography (3.5 × 25 cm2, acetone:water) to yield nine fractions (20, 30, 40, 50, 60, 70, 80, 90 and 100% acetone mixtures). The fractions eluted with 70% and 80% acetone–water (160 mg) were combined and subjected to further purification by gradient preparative HPLC using a reversed-phase column (Phenomenex Luna 10u C18(2) 100A (Phenomenex Torrance, CA, USA), 21.2 × 250 mm2, 10 μm; 10 ml min−1, 45% MeCN/H2O over 10 min, 45–50% MeCN/H2O over 10 min, 50% MeCN/H2O over 10 min, 50–100% MeCN/H2O over 20 min, 100% MeCN over 20 min) to afford compounds 1 (9.7 mg), 2 (1.9 mg), 3 (2.4 mg) and 5 (12.9 mg). The 90% (183.2 mg) and 100% (57.7 mg) acetone–water fractions were also subjected to the purification by gradient HPLC (Phenomenex Luna C18 preparative, 10 ml min−1, 50% MeCN/H2O over 10 min, 50–60% MeCN/H2O over 10 min, 60% MeCN/H2O over 20 min, 60–100% MeCN/H2O over 10 min, 100% MeCN over 20 min) to yield compounds 4 (35.1 mg), 5 (1.6 mg) and 6 (3.7 mg).
Saccharosporone A (1)
Yellow amorphous solid; [α]D −103.2 (c 0.3, MeOH); UV (MeOH) λmax (log ɛ) 215 nm (4.05), 245 nm (3.87), 273 nm (3.91), 421 nm (3.52); IR νmax (ATR) 3474, 2947, 1632, 1611, 1455, 1284, 1237, 1065 cm−1; for NMR data, see Table 1; ESIMS [M+Na]+ m/z 351; ESIMS [M+H]+ m/z 329; HRESITOFMS [M−H]− m/z 327.1231 (calcd for C19H19O5, 327.1232).
Saccharosporone B (2)
Yellow amorphous solid; [α]D −87.3 (c 0.03, MeOH); UV (MeOH) λmax (log ɛ) 215 nm (4.03), 245 nm (3.83), 273 nm (3.82), 421 nm (3.49); IR νmax (ATR) 3470, 2925, 1635, 1612, 1457, 1288, 1267, 1243, 1038 cm−1; for NMR data, see Table 1; ESIMS [M−H2O+Na]+ m/z 349; HRESITOFMS [M−H2O+Na]+ m/z 349.1047 (calcd for C19H18O5Na, 349.1052).
Saccharosporone C (3)
Yellow amorphous solid; [α]D −20.1 (c 0.07, MeOH); UV (MeOH) λmax (log ɛ) 222 nm (4.10), 259 nm (3.93), 313 nm (3.48); IR νmax (ATR) 3402, 2952, 2923, 1703, 1665, 1588, 1291, 1038 cm−1; for NMR data, see Table 1; ESIMS [M+Na]+ m/z 355; HRESITOFMS [M+H]+ m/z 333.1697 (calcd for C19H25O5, 333.1702).
Biological assays
Antimalarial activity against P. falciparum K1 was evaluated by the microculture radioisotope technique.10 The IC50 value of a standard antimalarial agent, dihydroartemisinin, was 0.001 μM. Antimicrobial assay against B. cereus was performed using the resazurin microplate technique.11 Vancomycin, used as a standard antibacterial drug, showed an MIC value of 0.69 μM. Growth inhibition against M. tuberculosis (H37Ra strain) was determined by a green fluorescent protein microplate assay.12 The MIC value of a standard drug, isoniazid, was 0.69 μM. A green fluorescent protein-based assay13 was used to determine cytotoxicity against Vero cell (African green monkey kidney fibroblasts), while antifungal activity against C. albicans and cytotoxic tests against cancer cell lines, including KB (human epidermoid carcinoma), MCF-7 (human breast cancer) and NCI-H187 (human small cell lung cancer), were performed using the resazurin microplate assay.14 The standard antifungal agent, amphotericin B, exhibited antifungal activity with an IC50 value of 0.13 μM, while ellipticine used as standard control for cyctotoxic assay against vero cell lines, displayed an IC50 value of 5.07 μM. Doxorubicin was used as positive controls for cytotoxic tests against KB, MCF-7 and NCI-H187 with respective IC50 values of 0.65, 16.3 and 0.1 μM. MIC and IC50 values >50 μg ml−1 were reported as inactive.
References
Krohn, K. & Rohr, J. Angucyclines: total syntheses, new structures, and biosynthetic studies of an emerging new class of antibiotics. Top. Curr. Chem. 188, 127–195 (1997).
Rohr, J. & Thiericke, R. Angucycline group antibiotics. Nat. Prod. Rep. 9, 103–137 (1992).
Bowie, J. H. & Johnson, A. W. The structure of ochromycinone. Tetrahedron Lett. 8, 1449–1452 (1967).
Taniguchi, M. et al. YM-181741, a novel benz[a]anthraquinone antibiotic with anti-Helicobacter pylori activity from Streptomyces sp. J. Antibiot. 55, 30–35 (2002).
Oka, M. et al. Chemical and biological properties of rubiginone, a complex of new antibiotics with vincristine-cytotoxicity potentiating activity. J. Antibiot. 43, 967–976 (1990).
Shigihara, Y. et al. 6-Deoxy-8-O-methylrabelomycin and 8-O-methylrabelomycin from a Streptomyces species. J. Antibiot. 41, 1260–1264 (1988).
Grabley, S. et al. Secondary metabolites by chemical screening. Part 19. SM 196 A and B, novel biologically active angucyclinones from Streptomyces sp. J. Antibiot. 44, 670–673 (1991).
Rickards, R. W. & Wu, J. P. Fujianmycins A and B, new benz[a]anthraquinone antibiotics from a Streptomyces species. J. Antibiot. 38, 513–515 (1985).
Pretsch, E., Simon, W., Seibl, J. & Clerc, T. Tables of Spectral Data for Structure Determination of Organic Compounds 2nd edn, I130 (Springer: Berlin, Germany, 1989).
Desjardins, R. E., Canfield, C. J., Haynes, J. D. & Chulay, J. D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16, 710–718 (1979).
Sarker, S. D., Nahar, L. & Kumarasamy, Y. Microtitre plate-based antibacterial assay incorporating resazurin as an indicator of cell growth, and its application in the in vitro antibacterial screening of phytochemicals. Methods 42, 321–324 (2007).
Collins, L. A., Torrero, M. N. & Franzblau, S. G. Green fluorescent protein reporter microplate assay for high-throughput screening of compounds against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 42, 344–347 (1998).
Changsen, C., Franzblau, S. G. & Palittapongarnpim, P. Improved green fluorescent protein reporter gene-based microplate screening for antituberculosis compounds by utilizing an acetamidase promoter. Antimicrob. Agents Chemother. 47, 3682–3687 (2003).
O'Brien, J., Wilson, I., Orton, T. & Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267, 5421–5426 (2000).
Acknowledgements
We are very grateful to Bioresources Research Network (BRN) of the National Center for Genetic Engineering and Biotechnology (BIOTEC) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Boonlarppradab, C., Suriyachadkun, C., Rachtawee, P. et al. Saccharosporones A, B and C, cytotoxic antimalarial angucyclinones from Saccharopolyspora sp. BCC 21906. J Antibiot 66, 305–309 (2013). https://doi.org/10.1038/ja.2013.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2013.16
