
Abstract
FD-594 is an unique pyrano[4′,3′:6,7]naphtho[1,2-b]xanthene polyketide with a trisaccharide of 2,6-dideoxysugars. In this study, we cloned the FD-594 biosynthetic gene cluster from the producer strain Streptomyces sp. TA-0256 to investigate its biosynthesis. The identified pnx gene cluster was 38 143 bp, consisting of 40 open reading frames, including a minimal PKS gene, TDP-olivose biosynthetic genes, two glycosyltransferase genes, two methyltransferase genes and many oxygenase/reductase genes. Most of these enzymes coded in the pnx cluster were reasonably assigned to a plausible biosynthetic pathway for FD-594, in which an unique ring opening process via Baeyer–Villiger-type oxidation catalyzed by a putative flavin adenine dinucleotide (FAD)-dependent monooxygenase, is speculated to lead to the unique xanthene structure. To clarify the involvement of pnx genes in the FD-594 biosynthesis, a glycosyltransferase, PnxGT2, and a methyltransferase, PnxMT2, were characterized enzymatically with the recombinant proteins expressed in Escherichia coli. As a result, PnxGT2 catalyzed the triple olivose transfers to the FD-594 aglycon with TDP-olivose as the glycosyl donor to afford triolivoside. Surprisingly, in the PnxGT2 enzymatic reaction, tetraolivoside and pentaolivoside were significantly detected along with the expected triolivoside. To our knowledge, PnxGT2 is the first contiguous oligosaccharide-forming glycosyltransferase in secondary metabolism. Furthermore, addition of PnxMT2 and S-adenosyl-L-methionine into the PnxGT2 reaction mixture afforded natural FD-594 to confirm that the PnxGT2 reaction product was the expected regiospecifically glycosylated compound. Consequently, the identified pnx gene cluster appears to be involved in FD-594 biosynthesis.
Similar content being viewed by others
Introduction
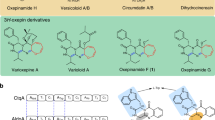
FD-594 is an aromatic polyketide antibiotic isolated from Streptomyces sp. TA-0256 with moderate cytotoxicity against several tumor cell lines and antibacterial activity.1 The polyketide aglycon of FD-594 consists of an unique pyrano[4′,3′:6,7]naphtho[1,2-b]xanthene skeleton, which is connected with a trisaccharide of 2,6-dideoxysugars, two D-olivoses and a D-oleandrose, through the phenolic oxygen at C12 of the F-ring (Figure 1).2 The absolute stereochemistry of FD-594 was determined using X-ray crystal structural analysis.3 We also reported the first instance of a unique solvent-dependent atropisomerism in a natural product from a structural study based on CD spectrometry and NMR spectroscopy.3 Feeding experiments with 13C-labeled sodium acetate revealed that the naphthoxanthene moiety is constructed via an unique ring opening at a quinone carbonyl group of a presumed benzo[a]naphthacenequinone intermediate.2 The ring-opened intermediate was then speculated to be recyclized and decarboxylated to the FD-594 skeleton. This unique biosynthetic feature prompted us to undertake detailed molecular analysis of FD-594 biosynthesis.

Structure of FD-594 and related aromatic polyketide antibiotics.
Many extensive biosynthetic studies of related bacterial aromatic polyketides have been conducted over many years, because this group includes many useful bioactive molecules, such as doxorubicin (antitumor), tetracycline (antibacterial), landomycin A (antitumor), pradimicin (antifungal) and gilvocarcin V (antitumor).4 All are formed by type II polyketide synthases (PKSs), which are comprised of two ketosynthases (KSα and KSβ) and an acyl carrier protein (ACP). The set of KSα/KSβ/ACP, designated as a minimal PKS, catalyzes the polyketide chain growth. The elongated linear polyketide chain is subsequently cyclized by a cyclase/aromatase, leading to the basic backbone of the aromatic polyketide, which is then modified by various tailoring enzymes, such as several types of oxygenase/reductase, methyltransferase and glycosyltrasferase enzymes. Accordingly, various combinations of the biosynthetic enzymes generate great structural diversity of aromatic polyketides in nature. Therefore, engineering of biosynthetic pathways by genetic manipulation and change of enzymatic activity can engender generation of novel molecules, which might serve as new bioactive compounds. To control the complex biosynthetic machinery, precise understanding of biosynthetic genes and enzymes is expected to be necessary.
Herein, we describe the cloning and sequencing of the FD-594 biosynthetic gene cluster from the producer Streptomyces sp. TA-0256 and propose a plausible biosynthetic pathway for FD-594 by bioinformatic analysis with the related aromatic polyketide biosynthetic gene clusters. Additionally, we establish the final steps of the biosynthesis with recombinant enzymes to clarify that the identified gene cluster is responsible for FD-594 biosynthesis.
Results and Discussion
Identification of the FD-594 biosynthetic gene cluster
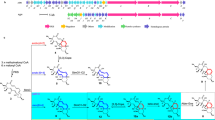
To identify the biosynthetic gene cluster for FD-594, we used unique gene probes for TDP-glucose-4,6-dehydratase (4,6-DH) and TDP-glucose-2,3-DH, which are generally involved in the biosyntheses for TDP-2,6-dideoxy sugars such as olivose and oleandrose.5, 6, 7 As a result of several PCR conditions with designed primers, the PCR with a set of primers for 2,3-DH gave a desired amplified DNA fragment. A pOJ446-based cosmid library with the randomly Sau3AI digested genomeic DNA of Streptomyces sp. TA-0256 was constructed and further screened using hybridization with the DIG-labeled probe DNA for the 2,3-DH gene. Consequently, three overlapped cosmids containing the 2,3-DH gene were obtained. Sequence analysis of these cosmids revealed an obvious type II PKS-containing gene cluster, which was sandwiched with contiguously conserved orfs in both Streptomyces coelicolor and Streptomyces avermitillis (Table 1). The gene cluster was 38 143 bp consisting of 40 orfs, including type II PKS and TDP-2,6-dideoxysugar biosynthetic genes (Table 1 and Figure 2). Although the precise cluster boundary was not determined, most of gene products were reasonably assigned for the FD-594 biosynthesis as describe below. We then named this gene cluster as the pnx cluster from the pyranonaphthaxanthene structure of FD-594.

FD-594 gene cluster. (a) Minimal PKS and associated genes; (b) redox tailoring genes; (c) TDP-olivose biosynthetic genes; (d) methyltransferase and glycosyltransferase genes; (e) regulatory and transporter genes and (f) unknown function genes or outside of the FD-594 biosynthetic gene cluster.
Conserved polyketide biosynthetic genes
The existence of a minimal PKS, PnxA (KSα), PnxB (KSβ) and PnxC (ACP), suggested that this gene cluster is responsible for bacterial aromatic polyketide biosynthesis. High sequence similarity (70–83% for the KSs and 55–75% for the ACP) to the related aromatic polyletide biosynthetic enzymes, such as pradimicin biosynthetic enzyme (Pdm),8, 9 fredericamycin biosynthetic enzyme (Fdm),10 lysolipin biosynthetic enzyme (Llp),11 griseorhodin biosynthetic enzyme (Grh),12 rubromycin biosynthetic enzyme (Rub)13 and benastatin biosynthetic enzyme (Ben),14 suggested that the identified minimal PKS (PnxA, PnxB and PnxC) seems to catalyze chain-elongation of a presumable biosynthetic intermediate for FD-594 (Table 1).
In the pnx cluster, three probable PKS-priming enzymes PnxU (so-called ketosynthase III, KSIII), PnxV (ACP) and PnxJ (acyltrasferase, AT) genes were identified.4, 15, 16, 17 Among the homologous enzymes, the R1128 biosynthetic enzymes ZhuH (KSIII), ZhuG (ACP) and ZhuC (AT) were well studied.15 ZhuH catalyzes the Claisen-type condensation between malonylated ZhuG and the acyl CoAs to afford the corresponding diketide intermediates on ZhuG.18 Then PnxU seems to catalyze the first condensation to synthesize the presumed butyrate starter unit as a priming KS. Indeed, PnxU and the minimal PKS PnxA/PnxB are expected to distinguish two ACPs, PnxV for a priming ACP and PnxC for an elongation ACP.19 ZhuC was reported to catalyze the malonyl trasfer reaction onto the priming ACP ZhuG,18 as well as removal of acetyl groups to edit the acetyl priming ZhuG.20 Consequently, PnxJ can be proposed to function as ZhuC in R1128 biosynthesis. In the initiation process, β-ketoreductase, DH and enoylreductase were recruited from bacterial fatty acid synthases in host strains, as suggested in the R1128 biosynthesis.21 However, 3-ketoacyl-ACP reductase FdmC, coded in the fdm cluster, was recently found to reduce the β-diketide intermediate synthesized using FdmS (KSIII) and FdmH (ACP).16 Therefore, 3-ketoacyl-ACP reductases PnxG, PnxW or PnxO1 might be responsible for the reduction in the chain initiation.
PnxG showed high similarity to 3-ketoacyl-ACP reductases such as PdmG (73%) and BenL (75%), which were confirmed to catalyze reduction of the C19 carbonyl group of the elongated polyketide chain.14 Consequently, PnxG seems to catalyze the same β-keto reduction at C19 in the FD-594 aglycon biosynthesis. PnxW was found as another 3-ketoacyl-ACP reductase showing less similarity to PdmG (65%). Because the C11 position should be reduced similarly in the process of polyketide backbone construction, PnxW might catalyze the β-keto reduction of the carbonyl group at C11 of the nascent polyketide chain.
Three putative cyclases, PnxD, PnxK and PnxL, are conserved, in the paradimicin (pdm) gene cluster as PdmD, PdmK and PdmL.22 Tang and co-workers expressed the minimal PKS PdmABC with the cyclases in S. coelicolor CH999 to investigate its functions.22 Although an additional highly conserved monooxygenase, PdmH, in this class of biosynthetic gene clusters was required, the pdmABCDHKL genes were sufficient for the biosynthesis of the pentangular polyphenol backbone of pradimicin A in the hetereologous host, indicating that the three cyclases, PdmDKL, are responsible for the cyclization and aromatization of the elongated linear polyketide chain. Consequently, the homologous PnxDKL is apparently responsible for the cyclization and aromatization to synthesize a presumable pentangular intermediate as shown in Figure 3. In fact, PnxD resembles the N-terminus of TcmN, which catalyzes the first and second cyclization of a linear polyketide chain in tetracenomycin biosynthesis.23, 24 Therefore, PnxD seems to catalyze the first two aldol-type cyclizations and dehydrations at C9–C14 and C7–C16 of a presumed linear intermediate. Because addition of TcmJ into the TcmKLM (minimal PKS) and TcmN system significantly increased tricyclic tetracenomycin F2 formation, TcmJ was found to support the formation of the first three rings.25, 26 Therefore, the function of the TcmJ homolog PnxL was presumed to be somehow involved in the stabilization of the accompanying cyclases and/or the conformation of the polyketide intermediate to afford the tricyclic polyketide. In view of the fact that PnxK homolog TcmI catalyzes the fourth ring formation in the tetracenomycin biosynthesis,26 PnxK is presumed to be responsible for the C–C bond formation at C4–C21 to give the tetracyclic intermediate. It remains unclear which enzyme in PdmABCDHKL is responsible for the fifth cyclization. An uncharacterized additional enzyme might be necessary to assist the fifth cyclization. Although a hydroxyl group at C11 is expected to be removed via dehydration in the proposed FD-594 biosynthesis, the responsible protein is also unclear. A unique enzyme encoded in the pnx cluster, such as PnxF or PnxM, might be responsible for the reaction.

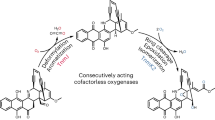
Proposed FD-594 biosynthetic pathway. A glycosyltransferase, PnxGT2, and a methyltransferase, PnxMT2, in the final two steps of the FD-594 biosynthesis were functionally characterized in this study. *Proteins outside of the gene cluster might catalyze reductions. ACP, acyl carrier protein; DH, dehydratase; ER, enoylreductase; KR, ketoreductase; KS, ketosynthase.
Redox enzymes for modification of the polyketide skeleton
Four small putative monooxygenases––PnxH, PnxI, PnxE1 and PnxE2––are encoded in the pnx cluster (Table 1). Among those, PnxH showed high similarity to a conserved monooxygenase PdmH (73%), which was confirmed to be responsible for quinone formation in the biosynthesis of the pentangular polyphenol backbone as described above.22 Furthermore, PnxH showed less similarity to a cofactor-independent monooxygenase SnoaB (46%) in nogalamycin biosynthesis.27 The crystal structure of SnoaB revealed that the completely conserved tryptophan residue in this family of antibiotic monooxygenases including ActVA-Orf6,28, 29 TcmH30 and AknX31 is catalytically important.32 Indeed, the corresponding tryptophan residue is conserved in PnxH, supporting its involvement in quinone formation. PnxI belongs to a family of antibiotic monooxygenase and also has a conserved tryptophan residue, indicating that PnxI also seems to catalyze the oxygenation of the para position of phenol moieties. Because PnxI homologs are conserved in the pdm and llP gene clusters, PnxI is apparently responsible for the oxygenation at C6, which should also be involved in pradimicin A and lysolipin biosynthesis (Figure 1).
The other putative monooxygenases PnxE1 and PnxE2 mutually share sequence identity and are highly conserved in structurally related aromatic polyketide biosynthetic gene clusters (Table 1). Recently, Shen and co-workers reported that PnxE1 and PnxE2 homologs FdmM and FdmM1 were responsible for the oxygenation at C6 and C8 of the polyketide intermediate by gene inactivation studies.33 However, this oxygenation activity is the same as that proposed for PnxH and PnxI described above. Therefore, at the moment, we propose that three of PnxH, PnxI, PnxE1 and PnxE2 might be responsible for the oxygenation at C6, C8 and C10. Further enzymological studies of these monooxygenases must be done to elucidate the oxidation chemistry in the biosynthesis of this class of highly oxidized aromatic polyketides.
Three cytochrome P450 monooxygenases––PnxO2, PnxO3 and PnxO5––are encoded in the gene cluster and are presumably involved in the hydroxylations of inactive carbon atoms. These proteins mutually share sequence identity and show similarity to PdmJ and PdmW in pradimicin biosynthesis. Heterologous expression of PdmJ and PdmW with PdmABCDGHKL in S. coelicolor CH999 clarified that both P450s are involved in the hydroxylation at two methylenes of dihydrobenzo[a]naphthacenequinone.34 Because the pdmABCDGHKL gene-containing heterologous strain somehow produced dihydrobenzo[a]naphthacenequinone via unknown dehydration and reduction at the bent reduced moiety of pentaangular polyketide intermediate,22 the resulting inactive two methylenes can be hydroxlylated by the two P450s PdmJ and PdmW. The same scenario is applicable for FD-594 biosynthesis. Pnx P450s would catalyze the hydroxylation of the corresponding moieties. PnxO3 shows the highest similarity to PdmW, which was proposed to catalyze the hydroxylation at C6 of the dihydrobenzo[a]naphthacenequinone core of pradimicin A. Both PnxO2 and PnxO5 show higher similarity to PdmJ (63% to both), which was confirmed unambiguously to catalyze the hydroxylation at C5 of the intermediate. Consequently, either of these seems to catalyze the hydroxylation at the corresponding site of the biosynthetic intermediate. The pnxO5 gene is located just upstream of a glycosyltransferase gene pnxGT1. Because cytochrome P450 homologous proteins DesVIII and AknT were reported to assist the folding of the accompanied glycosyltransferases DesVII and AknS in the biosyntheses of methymicin–pikromycin and aclacinomycin A, respectively,35, 36, 37 PnxO5 might function as a similar helper protein for PnxGT1.
Two FAD-dependent oxygenases, PnxO4 and PnxO6, are encoded in the gene cluster. PnxO6 shows similarity to PokO2 (46%) in polyketomycin biosynthesis and MtmOII (45%) in mithramycin biosynthesis.38, 39 The gene inactivation studies of pokO2 and mtmOII suggested that PokO2 and MtmOII catalyze the specific epoxidation reaction of tetracyclic aromatic polyketide intermediates. Consequently, a similar epoxidation reaction can be speculated to be catalyzed by PnxO6 before the unique ring opening process shown in Figure 3. PnxO4 shows similarity to RubP (58%) and RubL (56%) in rubromycin biosynthesis; GrhO5 (54%), GrhO8 (58%) and GrhO9 (56%) in griseorhodin biosynthesis and TcmG (57%) in tetracenomycin biosynthesis. Inactivation of the grhO5 gene in the griseorhodin biosynthetic gene cluster in a heterologous expression system of the whole grh gene cluster in Streptomyces albus generated collinone production.14 Therefore, GrhO5 was proposed as a key enzyme in the elaboration of the unique spiroketal formation. Consequently, PnxO4 is presumed to catalyze the Baeyer–Villiger-type oxidation to cleave the C–C bond of the presumed epoxide intermediate formed by PnxO6. The resulting lactone intermediate might be hydrolyzed, with subsequent rotation and recyclization via the ring opening of the epoxide, followed by decarboxylation, to afford the FD-594 aglycon. Two putative hydrolases––PnxN and PnxP––are specifically encoded in the pnx cluster and might be responsible for hydrolysis in the ring opening process.
PnxO1 shows medium similarity to many NADP(H)-dependent, short-chain dehydrogenases/reductases, including 3-ketoacyl-ACP reductases such as ActIII (51%) and med-ORF6 (52%), which are responsible for the C9 keto reduction in actinorhodin biosynthesis and medermycin biosynthesis.40, 41 Consequently, PnxO1 might catalyze the β-keto reduction of C11 of the nascent polyketide chain instead of PnxW described above. As another possibility, PnxO1 might catalyze the reduction at the C25 position and the subsequent lactone ring formation, which is a necessary transformation in FD-594 biosynthesis.
PnxO7 shows medium similarity to many NAD-dependent 3-hydroxyisobutyrate dehydrogenases; it is conserved in the llp gene cluster as the llpS gene (67%). Accordingly, PnxO7 seems to catalyze the oxidation of a hydroxyl group to give a carbonyl group, but such transformation is not expected. PnxO7 and LlpS might have the same function in each biosynthesis.
PnxO8 shows medium similarity to a putative monooxygenase of unknown function, which is conserved in the llp gene cluster (LlpV, 70%) and fdm gene cluster (FdmL, 61%). Consequently, this type of protein is apparently related to aromatic polyketide biosynthesis, although its function remains unclear at present.
Biosynthetic genes for TDP-olivose
A set of TDP-olivose biosynthetic genes was easily identified in the pnx cluster based on the other TDP-2,6-dideoxysugar biosynthetic genes.42 TDP-glucose synthase PnxS1, TDP-glucose-4,6-DH PnxS2, NDP-hexose-2,3-DH PnxS3, NDP-hexose-3-ketoreductase PnxS4 and NDP-hexose-4-ketoreductase appeared responsible for TDP-olivose formation from D-glucose-1-phosphate. TDP-olivose is expected to be supplied as the glycosyl donor for the triple glycosylations in the construction of the trisaccharide moiety of FD-594.
Glycosyltransferases and methyltransferases
Only two glycosyltransferases––PnxGT1 and PnxGT2––were identified in the cluster, although three glycoside bonds must be generated in FD-594 biosynthesis. Therefore, one of these glycosyltransferases seems to work at least twice in the olivosyl transfer. PnxGT1 belongs to family 28 of glycosyltransferases and shows medium similarity to PdmS (54%) in pradimicin biosynthesis, in which PdmS was presumed to attach a sugar on the D ring of the naphthacenequinone of the pradimicin aglycon or its monoglycoside. However, FD-594 contains no glycoside at the corresponding moiety, indicating that PnxGT1 could be involved in the other site of glycosylation. In addition, PnxGT1 showed high similarity to N-glycosyltransferase AtG in indolocarbazole antibiotic AT2433 biosynthesis.43 Therefore, PnxGT1 was presumed to catalyze the first olivosylation at the phenolic hydroxyl group at C12 of the FD-594 aglycon. PnxGT2 belongs to glycosyltransferase family 28 and shows similarity to many O-glycosyltransferases, including LanGT1 (62%), LanGT3 (55%) and LanGT4 (47%) in landomycin biosynthesis and AtG1 (50%) in AT2433 biosynthesis. Because LanGT1 and LanGT4 were confirmed to be used iteratively in landomycin biosynthesis,44 PnxGT2 was proposed to similarly catalyze two glycosylations to complete the trisaccharide formation in FD-594 biosynthesis.
Two putative methyltrasferases, PnxMT1 and PnxMT2, were identified in the pnx cluster, indicating that one is responsible for the methylation of the phenolic hydroxyl group of the polyketide aglycon; another methyltransferase is expected to catalyze the methylation of the terminal olivose moiety into oleandrose. Because PnxMT1 shows medium similarity to N-methyltransferase AtM1 (54%) in indolocarbazole antibiotic AT2433 biosynthesis,43 PnxMT1 was presumed to catalyze the methylation of the phenolic hydroxyl group of the FD-594 aglycon. On the other hand, PnxMT2 shows medium similarity to the FkbM family of O-methyltransferases45 (>50%), suggesting that PnxMT2 catalyzes methylation of the terminal olivose.
Regulators, transporters and others
Two transcriptional regulators––PnxR1 and PnxR2––are encoded in the gene cluster. PnxR1 belongs to the MarR family of transcriptional regulators, which repress expression of the corresponding operons by binding to the central regulatory region for transcription that is inducible by structurally unrelated antibiotics such as tetracycline and chloramphenicol.46 PnxR1 might control the expression of the transporter gene pnxT by response to the accumulation of FD-594. PnxR2 shows similarity to the streptomyces antibiotic regulatory protein family of transcriptional regulators (50%), which are known to act as a transcriptional activators.47 Similar streptomyces antibiotic regulatory protein family transcriptional regulators are encoded in the related polyketide biosynthetic gene clusters. Inactivation of FdmR1 in the fdm cluster abolished fredericamycin biosynthesis, supporting its function as the transcriptional activator.48 The expression and modification of PnxR2 was apparently necessary for the initiation of transcription of the pnx cluster.
PnxT shows similarity to transmembrane efflux proteins, indicating that PnxT carries FD-594 outside of the membrane as a transporter.
PnxN shows similarity to N-acyl homoserine lactone lactonase and metallo-β-lactamase (>60%). FD-594 has a lactone moiety, which might be hydrolyzed by this putative hydrolase PnxN. However, the exact functions of such enzymes remain unclear. Alternatively, PnxN might function as a quorum-quencher by hydrolysis of a quorum-sensing molecule such as A-factor, which triggers aerial mycelium formation and secondary metabolism in many streptomyces.49
PnxP shows similarity to another family of hydrolases, including amidohydrolase and organophosphate acid anhydrase, which are highly conserved in Streptomyces. As described above, in the proposed ring opening reaction of the presumed polyketide biosynthetic intermediate, a certain hydrolysis of the lactone ring was speculated. Consequently, PnxN or PnxP might be involved in such a hydrolysis reaction to construct the FD-594 aglycon. However, because these homologous proteins are not conserved in lysolipin biosynthesis, for which a similar ring opening and recycling of the polyketide was inferred, PnxN and PnxP might not be involved directly in FD-594 biosynthesis. Because a discrete β-lactamase-type thioesterase was established to catalyze the release of the polyketide thioester on the iterative type I PKS,50 PnxN or PnxP might catalyze such a polyketide release from the minimal PKS PnxABC.
PnxF showed medium similarity (40–60%) to hypothetical proteins of unknown function that are highly conserved in Actinomycetes, but not in the related aromatic biosynthetic gene clusters. Consequently, PnxF seems not to be involved in FD-594 biosynthesis.
The putative pnxM gene product is a small protein, which has no putative conserved domains, suggesting no relation to FD-594 biosynthesis.
Functional analysis of glycosyltransferase, PnxGT2, and methyltransferase, PnxMT2
Genetic manipulation of the FD-594 producer Streptomyces sp. TA-0256 to inactivate pnx genes has never succeeded to date. Therefore, we attempted to characterize the enzymatic function of the putative Pnx proteins using recombinant proteins expressed in E. coli. As we proposed the biosynthetic functions, two glycosyltransferases, PnxGT1 and PnxGT2, were expected to catalyze the glycosylations of the FD-594 aglycon. Unfortunately, soluble PnxGT1 was not obtained at all with several expression conditions. On the other hand, soluble PnxGT2 protein was obtained when it was coexpressed with the molecular chaperons GroES and GroEL at lower temperature. Recombinant PnxGT2 was also purified successfully with standard Ni-binding affinity chromatography. The obtained pure PnxGT2 was reacted with FD-594 aglycon and TDP-olivose. It is particularly interesting that large amounts of new compounds were generated in the PnxGT2 enzymatic reaction (Figure 4a). LC–ESIMS analysis of the major products revealed that these compounds corresponded to the triolivoside, tetraolivoside and pentaolivoside of the FD-594 aglycon with conversion yields of 17, 18 and 4%, respectively, based on the remaining starting material. In this analysis, a trace amount of disaccharide, hexasaccharide and heptasaccharide were also detected.

HPLC traces of PnxGT2 and PnxMT2 enzymatic reaction products. LC–ESI–MS analysis was performed using LCQ (Finnigan) coupled with Nanospace HPLC (Shiseido, Yokohama, Japan) with a standard ODS column. The elution was made using 10% CH3CN(aq.) for 10 min, followed by a linear gradient of 40–100% CH3CN(aq.) for 30 min at a flow rate of 50 μl min–1. See details in Supplementary Information. (a) PnxGT2 with aglycon and TDP-olivose; (b) PnxGT2 and PnxMT2 with aglycon, TDP-olivose and S-adenosyl-L-methionine; (c) PnxMT2 with S-adenosyl-L-methionine after inactivation of PnxGT2 in a; (d) a half amount of B plus authentic FD-594; (e) aglycon and TDP-olivose (no enzyme) as control. The LC traces of LC–ESI–MS analysis are similar to the HPLC-UV traces; the raw data were omitted. Instead, the m/z values of the corresponding peaks in the negative mode ESI–MS analysis are shown in these traces.
The contiguous iterative glycosylation ability of PnxGT2 is an intriguing issue of enzymatic chemistry; how does PnxGT2 recognize substrates and controls the number of the glycosylations? Because triolivoside and tetraolivoside were the main products, PnxGT2 appears to control the number of sugars with the limited size of the active site. In contrast, monosaccharide and disaccharide were almost not detected in the PnxGT2 reaction, even though the starting FD-594 aglycon remained in the reaction mixture, suggesting that triolivoside formation is much faster than monosaccharide and disaccharide formation. This might imply that PnxGT2 prefers mono-olivosides and diolivosides as substrates to the FD-594 aglycon. In landomycin A biosynthesis, LanGT1 and LanGT4 are used twice to attach two D-olivoses and two L-rhodinoses, respectively.51 The recognition mechanism of these iterative glycosyltransferase remains unclear at present; more detailed biochemical studies are necessary to obtain sufficient insight into the enzymology to engineer such glycosyltransferases to create diverse glycosides using biosynthetic technology. The function of PnxGT1 is unclear because PnxGT2 catalyzes triolivoside formation from the FD-594 aglycon. Because the second and third glycosylation catalyzed by PnxGT2 were apparently the preferential enzymatic reaction, PnxGT1 might catalyze the first glycosylation of olivose to the FD-594 aglycon. Otherwise, because no preferential glycosyl donor exists in the producer strain, PnxGT1 might not be involved in the biosynthesis of FD-594. Enzymatic reaction analysis of PnxGT1 with several glycosyl donors could provide some clue to elucidate the function.
Regarding oleandrose formation in FD-594 biosynthesis, OleY methyltransferase reportedly recognizes L-olivosyl-erythronolide B and catalyzes the methylation of the hydroxyl group at C3 of olivose in oleandomycin biosynthesis.52 Glycosyltransferase OleG2 catalyzes the olivosyl transfer to erythronolide B and cannot transfer L-oleandrose to the aglycon, supporting the post-glycosyltransferase methylation pathway for the oleandrose moiety. Because a similar scenario for the methylation reaction to form the oleandrose moiety is apparently applicable to the biosynthesis of FD-594, we next attempted to convert the PnxGT2 enzymatic reaction product into FD-594 using a putative methyltransferase, PnxMT2.
PnxMT2 was easily expressed in E. coli, and its pure form was prepared. PnxMT2 and a probable methyl donor S-adenosyl-L-methionine were then added to the PnxGT2 reaction mixture. As a result, the PnxGT2 reaction products were reduced, and FD-594 was generated with a conversion yield of 24% based on the remaining FD-594 aglycon (Figures 4b and d). To confirm that PnxMT2 catalyzes the methylation of the triolivoside generated by PnxGT2, the reaction mixture of PnxGT2 with FD-594 aglycon and TDP-olivose was heat-treated; then, fresh PnxMT2 and S-adenosyl-L-methionine were added. As we anticipated, a similar FD-594 formation was detected, which indicates that PnxMT2 recognizes the triolivoside and catalyzes the methylation to complete FD-594 biosynthesis. Consequently, these results verified clearly that in the final steps of FD-594 biosynthesis, a glycosyltransferase PnxGT2 catalyzes three olivose transfers to the FD-594 aglycon with TDP-olivose and then a methyltransferase, PnxMT2, attaches the methyl group at the terminus of the triolivoside. These results support that the identified pnx gene cluster should be responsible for FD-594 biosynthesis.
A major accompanied product in the stepwise reaction of PnxGT2 and PnxMT2 was found to be a methylated tetraolivoside of the FD-594 aglycon (Figure 4c). This result demonstrates that PnxMT2 recognized both triolivoside and tetraolivoside in the solution of the PnxGT2 enzymatic reaction and then transferred a methyl group to the hydroxyl group of C3 in olivose. In contrast, for the one-pot enzymatic reaction of PnxGT2 and PnxMT2, FD-594 was the major product rather than tetraolivoside, indicating that the PnxMT2 reaction with triolivoside is faster than the attachment of the fourth olivose to the triolivoside by PnxGT2. These results could imply that the existence of the methoxy group at C3 of the terminal sugar of FD-594 inhibits the additional glycosylation. This story for the completion of the biosynthesis agreed well with the explanation that only FD-594 was accumulated in the culture of the producer strain.
Overall biosynthesis of FD-594 (Figure 3)
In this study, we identified a structurally unique FD-594 biosynthetic gene cluster with the NDP-hexsose-2,3-DH gene as a probe. Most of the pnx gene products were functionally assigned for FD-594 biosynthesis, as described above. An initiation module consisting of PnxU (KSIII) and PnxV (ACP) was proposed to be involved in the preparation of the butyryl starter unit for the minimal PKS, PnxABC. Then, PnxABC extends the polyketide chain with 12-malonyl-CoA as extender units. The elongated linear polyketide chain is then cyclized and aromatized by three cyclases, PnxDKL, via five aldol-type condensations and six dehydrations to give a pentangular polyketide backbone. During the cyclization process, two carbonyl groups would be reduced by 3-keto-ACP ketoreductases PnxG, PnxW or PnxO1. The resulting polyketide biosynthetic intermediate is oxidized by four antibiotic monooxygenases––PnxH, PnxI, PnxE1 and PnxE2––and two of three cytochrome P450s—PnxO2, PnxO3 or PnxO5. The butyrate starter moiety should be reduced by a reductase, such as PnxO1, to construct the terminal lactone moiety. In the final modification to construct the FD-594 aglycon, two FAD-dependent monooxygenases, PnxO4 and O6, are responsible for ring opening, hydrolysis, recycling and decarboxylation to produce the unique xanthene moiety. Two hydrolases, PnxN and PnxP, might be involved in the hydrolysis reaction in the process.
PnxS1, S2, S3, S4 and S5 would synthesize TDP-olivose from glucose-1-phosphate. Then, PnxGT2 transfers the olivose from TDP-olivose to synthesize the triolivoside. A methyltransferase, PnxMT1, would catalyze the methylation of the phenolic hydroxyl group at C8 of the FD-594 aglycon. Finally, PnxMT2 completes FD-594 biosynthesis by attaching the methyl group on the terminal olivose.
In the proposed biosynthetic pathway, the ring-opening and ring-closing process of the FD-594 aglycon is an intriguing transformation. Detailed functional analyses of the Pnx proteins must be undertaken to provide further insight into this post-PKS process to elucidate the complex biosynthetic pathway for FD-594.
In conclusion, we identified the biosynthetic gene cluster for the highly oxidized aromatic polyketide FD-594 and propose a plausible biosynthetic pathway with coded proteins in the cluster. Although the precise functions of many Pnx proteins remain unclear at present, the present genetic information should provide important clues that might eventually elucidate this class of angucyclic polyketide biosynthesis. Especially, the unique xanthene formation involved in the FD-594 biosynthesis would be an intriguing issue to be solved in the future. Indeed, more detailed biochemical studies of characterized PnxGT2 and PnxMT2 are necessary to elucidate these enzymes and eventually to support engineered biosyntheses for creation of novel compounds.
Methods
Bacterial strains and growth conditions
Streptomyces sp. TA-0256 was used as the FD-594 producer strain and source of the FD-594 biosynthetic genes (pnx). Streptomyces sp. TA-0256 was maintained on International Streptomyces Project 2 (ISP2) (0.4% yeast extract, 1% malt extract, 0.4% glucose, pH 7.3) agar medium (2.0% agar, 28 °C, 3 days); the spores were used for a seed culture in ISP2 liquid medium (28 °C, 200 r.p.m., 24 h). Genomic DNA was extracted from the cultured cells in 100 ml of ISP2 liquid medium containing 1.5% glycine (28 °C, 200 r.p.m., 2 days) according to standard method described in Practical Streptomyces Genetics.53
For plasmid preparation, Escherichia coli DH5α was used routinely. E. coli XL-1 Blue MRF’ was used for cosmid manipulations. E. coli BL21(DE3) was used for the pnxGT2 and pnxMT2 gene expressions. Luria–Bertani medium supplemented with standard amounts of antibiotic as required (50 μg ml−1 ampicillin for LITMUS28 and pT7-Blue) was used for the culture of E. coli. Genetic manipulation in E. coli was conducted according to a standard procedure.54
Identification of the FD-594 biosynthetic gene (pnx) cluster
A partial NDP-hexose-2,3-dehydratase (2,3-DH) gene was first amplified by PCR with a set of primers, 2,3-DH3: 5′-GAYGTSCTCCAGTCCGAGCA-3′ and 2,3-DH4C: 5′-GAASCGSCCGCCCTCCTCSGA-3′ (S, a mixture of C and G; Y, a mixture of C and T). DIG-labeled DNA for the 2,3-DH gene was utilized for screening of a pOJ446-based cosmid library. Consequently, three positive clones (c594A, c594B and c594C) were obtained. Among those, cosmid c594B was sequenced randomly using a shotgun sequence method on double-stranded DNA templates with more than 10-fold coverage and minimum three times each portion of the DNA sequence (Shimadzu Biotech, Kyoto, Japan). All gaps between the obtained contigs were separately sequenced. As a result, a 43 026-bp DNA sequence containing a type II PKS gene cluster was determined. The open reading frames (orfs) were determined using FramePlot analysis (http://www.nih.go.jp/~jun/cgi-bin/frameplot.pl)55 and a BLAST homology search using the NCBI BLAST server. The determined DNA sequence data of the pnx gene cluster in Streptomyces sp. TA-0256 has been deposited to the DDBJ databases under accession number AB469194.
Enzymatic characterization of PnxGT2
The pnxGT2 gene was amplified with a set of primers, GT2F: 5′-GGAAGGCATATGAGAATCCTCATGACG-3′ and GT2R: 5′-GCTGCTCGGATCCGCGCCGGTGCGTGCC-3′, and was cloned with an expression plasmid pColdI (Takara Bio, Shiga, Japan), resulting in pnxGT2/pColdI. pnxGT2 was coexpressed with molecular chaperones GroES and GroEL (pREP4-groESL) in BL21 (DE3).56 The E. coli harboring pnxGT2/pColdI/pREP4-groESL was cultured in Luria–Bertani medium with 50 μg ml–1 ampicillin and 30 μg ml−1 kanamycin at 37 °C by OD600 0.7 and a final 0.2 mM isopropyl β-D-thiogalactoside was then added for induction of overexpression at 24 °C.
The wet cells were suspended in 50 mM Tris buffer (pH 7.5) containing 10% glycerol, 1 mM MgCl2 and 1 mM MnCl2 (buffer A) and were disrupted by sonication. PnxGT2 was purified with a column of His-bind resin (Novagen, Darmstadt, Germany). To remove imidazole, the purified fraction was passed through a 5-ml desalting column (HiTrap; GE Healthcare, Buckinghamshire, UK) with buffer A. Then the PnxGT2-containing solution was concentrated with an ultrafiltration membrane (10 000 MW, Vivaspin 20; Sartorius AG, Gottingen, Germany). The PnxGT2 concentration was estimated using Lowry methods with bovine serum albumin as a standard. Usually, 1 ml of pure PnxGT2 (8.2 mg ml–1, 1.2 mM) was obtained from 6.6 g of wet cells.
The FD-594 aglycon was prepared using acid hydrolysis of FD-594, according to procedures described in a previous report.2 TDP-D-olivose was chemically synthesized according to the previous method.57 The PnxGT2 enzymatic solution consisted of 0.5 mM FD-594 aglycon, 2.0 mM TDP-olivose, 2.5% dimethylsulfoxide and 70 μM of pure PnxGT2 (total 100 μl). The PnxGT2 enzymatic reaction was performed at 28 °C for 24 h; then the reaction products were extracted three times with 100 μl ethyl acetate after acidification of the reaction mixture to pH 5.0. After removal of the solvent, the residue was dissolved with 100 μl methanol for HPLC (UV or photo-diode-array) and LC–ESI–MS analysis (see Supplementary Information).
Enzymatic characterization of PnxMT2
The pnxMT2 gene was amplified using a set of primers, MT2-F: 5′-GGAGAGATCATATGACCGGACC-3′ and MT2-R: 5′-CAGGGGGGTACCGACGCTC-3′, and was cloned with an expression plasmid, pColdI, to give pnxMT2/pColdI. The E. coli harboring pnxMT2/pColdI was cultured in Luria–Bertani medium with 50 μg ml–1 ampicillin at 37 °C by OD600 0.7 and a final 0.2 mM isopropyl β-D-thiogalactoside was then added for induction of overexpression. PnxMT2 was purified using the same method as for PnxGT2. Usually, 1 ml of pure PnxMT2 (8.8 mg ml–1, 0.31 mM) was obtained from 1.0 g of wet cells.
The PnxMT2 enzymatic reaction coupled with the PnxGT2 enzymatic reaction was conducted as follows. The enzymatic solution consisted of 0.2 mM FD-594 aglycon, 10 mM TDP-olivose, 5 mM S-adenosyl-L-methionine, 2% dimethylsulfoxide, 25 μM PnxGT2 and 90 μM PnxMT2 (total 100 μl). The enzymatic reaction was performed at 28 °C for 14 h. The analytical conditions for the enzymatic reaction were identical to the method described above for the PnxGT2 enzymatic reaction. For the stepwise enzymatic reaction analysis, the first reacted enzyme was heat-inactivated at 80 °C for 5 min. The PnxGT2 reaction was first conducted in 0.2 mM FD-594 aglycon, 10 mM TDP-olivose, 2% dimethylsulfoxide and 25 μM PnxGT2 (total 100 μl) at 28 °C for 18 h. Then, the mixture was heat-treated at 80 °C for 5 min, a final 3.3 mM of S-adenosyl-L-methionine and 75 μM of PnxMT2 were added to the mixture (total 150 μl), and the reaction was continued for 24 h.
References
Qiao, Y. F. et al. Isolation and characterization of a new pyrano[4′,3′:6,7]naphtho[1,2-b]xanthene antibiotic FD-594. J. Antibiot. 51, 282–287 (1998).
Kondo, K., Eguchi, T., Kakinuma, K., Mizoue, K. & Qiao, Y. F. Structure and biosynthesis of FD-594; a new antitumor antibiotic. J. Antibiot. 51, 288–295 (1998).
Eguchi, T. et al. Unique solvent-dependent atropisomerism of a novel cytotoxic naphthoxanthene antibiotic FD-594. J. Org. Chem. 64, 5371–5376 (1999).
Hertweck, C., Luzhetskyy, A., Rebets, Y. & Bechthold, A. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 24, 162–190 (2007).
He, X. M. & Liu, H. W Formation of unusual sugars: mechanistic studies and biosynthetic applications. Annu. Rev. Biochem. 71, 701–754 (2002).
Ogasawara, Y. et al. Cloning, sequencing, and functional analysis of the biosynthetic gene cluster of macrolactam antibiotic vicenistatin in Streptomyces halstedii. Chem. Biol. 11, 79–86 (2004).
Decker, H. et al. A general approach for cloning and characterizing dNDP-glucose dehydratase genes from actinomycetes. FEMS Microbiol. Lett. 141, 195–201 (1996).
Dairi, T., Hamano, Y., Igarashi, Y., Furumai, T. & Oki, T. Cloning and nucleotide sequence of the putative polyketide synthase genes for pradimicin biosynthesis from Actinomadura hibisca. Biosci. Biotechnol. Biochem. 61, 1445–1453 (1997).
Kim, B. C., Lee, J. M., Ahn, J. S. & Kim, B. S Cloning, sequencing, and characterization of the pradimicin biosynthetic gene cluster of Actinomadura hibisca P157-2. J. Microbiol. Biotech. 17, 830–839 (2007).
Wendt-Pienkowski, E. et al. Cloning, sequencing, analysis and heterologous expression of the fredericamycin biosynthetic gene cluster from Streptomyces griseus. J. Am. Chem. Soc. 127, 16442–16452 (2005).
Lopez, P. et al. Isolation of the lysolipin gene cluster of Streptomyces tendae Tu 4042. Gene 461, 5–14 (2010).
Li, A. & Piel, J. Gene cluster from a marine Streptomyces encoding the biosynthesis of the aromatic spiroketal polyketide griseorhodin A. Chem. Biol. 9, 1017–1026 (2002).
Saito, H., Bruenker, P., Martin, R. & Minas, W. Streptomyces collinus DSM2012 rubromycin biosynthesis gene cluster Accession number AF293355 (2002).
Lackner, G. et al. Biosynthesis of pentangular polyphenols: deductions from the benastatin and griseorhodin pathways. J. Am. Chem. Soc. 129, 9306–9312 (2007).
Das, A. & Khosla, C. Biosynthesis of aromatic polyketides in bacteria. Acc. Chem. Res. 42, 631–639 (2009).
Das, A., Szu, P.- H., Fitzgerald, J. T. & Khosla, C. Mechanism and engineering of polyketide chain initiation in fredericamycin biosynthesis. J. Am. Chem. Soc. 132, 8831–8833 (2010).
Xu, Z., Metsa-Ketela, M. & Hertweck, C. Ketosynthase III as a gateway to engineering the biosynthesis of antitumoral benastatin derivatives. J. Biotechnol. 140, 107–113 (2009).
Meadows, E. S. & Khosla, C. In vitro reconstitution and analysis of the chain initiating enzymes of the R1128 polyketide synthase. Biochemistry 40, 14855–14861 (2001).
Tang, Y., Lee, T. S., Kobayashi, S. & Khosla, C. Ketosynthases in the initiation and elongation modules of aromatic polyketide synthases have orthogonal acyl carrier protein specificity. Biochemistry 42, 6588–6595 (2003).
Tang, Y., Koppisch, A. T. & Khosla, C. The acyltransferase homologue from the initiation module of the R1128 polyketide synthase is an acyl-ACP thioesterase that edits acetyl primer units. Biochemistry 43, 9546–9555 (2004).
Tang, Y., Lee, H. Y., Kim, C. Y., Mathews, I. & Khosla, C. Structural and functional studies of SCO1815: a beta-ketoacyl-acyl carrier protein reductase from Streptomyces coelicolor A3(2). Biochemistry 45, 14085–14093 (2006).
Zhan, J., Watanabe, K. & Tang, Y. Synergistic actions of a monooxygenase and cyclases in aromatic polyketide biosynthesis. ChemBioChem 9, 1710–1715 (2008).
Shen, B. & Hutchinson, C. R. Deciphering the mechanism for the assembly of aromatic polyketides by a bacterial polyketide synthase. Proc. Natl Acad. Sci. USA 93, 6600–6604 (1996).
Ames, B. D. et al. Crystal structure and functional analysis of tetracenomycin ARO/CYC: implications for cyclization specificity of aromatic polyketides. Proc. Natl Acad. Sci. USA 105, 5349–5354 (2008).
McDaniel, R., Ebert-Khosla, S., Hopwood, D. A. & Khosla, C. Engineered biosynthesis of novel polyketides. Science 262, 1546–1550 (1993).
Shen, B. & Hutchinson, C. R. Tetracenomycin F2 cyclase: intramolecular aldol condensation in the biosynthesis of tetracenomycin C in Streptomyces glaucescens. Biochemistry 32, 11149–11154 (1993).
Torkkell, S. et al. The entire nogalamycin biosynthetic gene cluster of Streptomyces nogalater: characterization of a 20-kb DNA region and generation of hybrid structures. Mol. Genet. Genomics 266, 276–288 (2001).
Kendrew, S. G., Hopwood, D. A. & Marsh, E. N. Identification of a monooxygenase from Streptomyces coelicolor A3(2) involved in biosynthesis of actinorhodin: purification and characterization of the recombinant enzyme. J. Bacteriol. 179, 4305–4310 (1997).
Sciara, G. et al. The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin biosynthesis. EMBO J. 22, 205–215 (2003).
Shen, B. & Hutchinson, C. R. Tetracenomycin F1 monooxygenase: oxidation of a naphthacenone to a naphthacenequinone in the biosynthesis of tetracenomycin C in Streptomyces glaucescens. Biochemistry 32, 6656–6663 (1993).
Chung, J. Y., Fujii, I., Harada, S., Sankawa, U. & Ebizuka, Y. Expression, purification, and characterization of AknX anthrone oxygenase, which is involved in aklavinone biosynthesis in Streptomyces galilaeus. J. Bacteriol. 184, 6115–6122 (2002).
Grocholski, T. et al. Crystal structure of the cofactor-independent monooxygenase SnoaB from Streptomyces nogalater: Implications for the reaction mechanism. Biochemistry 49, 934–944 (2010).
Chen, Y., Wendt-Pienkoski, E., Rajski, S. R. & Shen, B. In vivo investigation of the roles of FdmM and FdmM1 in fredericamycin biosynthesis unveiling a new family of oxygenases. J. Biol. Chem. 284, 24735–24743 (2009).
Zhan, J., Qiao, K. & Tang, Y. Investigation of tailoring modifications in pradimicin biosynthesis. ChemBioChem 10, 1447–1452 (2009).
Borisova, S. A., Zhao, L., Melancon, I. C., Kao, C. L. & Liu, H. W Characterization of the glycosyltransferase activity of desVII: analysis of and implications for the biosynthesis of macrolide antibiotics. J. Am. Chem. Soc. 126, 6534–6535 (2004).
Borisova, S. A. & Liu, H. W. Characterization of the glycosyltransferase DesVII and its auxiliary partner protein DesVIII in the methymycin/pikromycin biosynthetic pathway. Biochemistry 49, 8071–8084 (2010).
Leimkuhler, C. et al. Characterization of rhodosaminyl transfer by the AknS/AknT glycosylation complex and its use in reconstituting the biosynthetic pathway of aclacinomycin A. J. Am. Chem. Soc. 129, 10546–10550 (2007).
Daum, M. et al. Organisation of the biosynthetic gene cluster and tailoring enzymes in the biosynthesis of the tetracyclic quinone glycoside antibiotic polyketomycin. ChemBioChem 10, 1073–1083 (2009).
Abdelfattah, M. S. & Rohr, J. Premithramycinone G, an early shunt product of the mithramycin biosynthetic pathway accumulated upon inactivation of oxygenase MtmOII. Angew. Chem. Int. Ed. Engl. 45, 5685–5689 (2006).
Hadfield, A. T., Limpkin, C., Teartasin, W., Simpson, T. J., Crosby, J. & Crump, M. P. The crystal structure of the actIII actinorhodin polyketide reductase: proposed mechanism for ACP and polyketide binding. Structure 12, 1865–1875 (2004).
Ichinose, K., Ozawa, M., Itou, K., Kunieda, K. & Ebizuka, Y. Cloning, sequencing and heterologous expression of the medermycin biosynthetic gene cluster of Streptomyces sp. AM-7161: towards comparative analysis of the benzoisochromanequinone gene clusters. Microbiology 149, 1633–1645 (2003).
Trefzer, A., Salas, J. A. & Bechthold, A. Genes and enzymes involved in deoxysugar biosynthesis in bacteria. Nat. Prod. Rep. 16, 283–299 (1999).
Gao, Q., Zhang, C., Blanchard, S. & Thorson, J. S. Deciphering indolocarbazole and enediyne aminodideoxypentose biosynthesis through comparative genomics: insights from the AT2433 biosynthetic locus. Chem. Biol. 13, 733–743 (2006).
Luzhetskyy, A. et al. Iteratively acting glycosyltransferases involved in the hexasaccharide biosynthesis of landomycin A. Chem. Biol. 12, 725–729 (2005).
Motamedi, H. et al. Characterization of methyltransferase and hydroxylase genes involved in the biosynthesis of the immunosuppressants FK506 and FK520. J. Bacteriol. 178, 5243–5248 (1996).
Seoane, A. S. & Levy, S. B. Characterization of MarR, the repressor of the multiple antibiotic resistance (mar) operon in Escherichia coli. J. Bacteriol. 177, 3414–3419 (1995).
Floriano, B. & Bibb, M. afsR is a pleiotropic but conditionally required regulatory gene for antibiotic production in Streptomyces coelicolor A3(2). Mol. Microbiol. 21, 385–396 (1996).
Chen, Y., Wendt-Pienkowski, E. & Shen, B. Identification and utility of FdmR1 as a Streptomyces antibiotic regulatory protein activator for fredericamycin production in Streptomyces griseus ATCC 49344 and heterologous hosts. J. Bacteriol. 190, 5587–5596 (2008).
Ohnishi, Y., Yamazaki, H., Kato, J. Y., Tomono, A. & Horinouchi, S. AdpA, a central transcriptional regulator in the A-factor regulatory cascade that leads to morphological development and secondary metabolism in Streptomyces griseus. Biosci. Biotechnol. Biochem. 69, 431–439 (2005).
Awakawa, T. et al. Physically discrete beta-lactamase-type thioesterase catalyzes product release in atrochrysone synthesis by iterative type I polyketide synthase. Chem. Biol. 16, 613–623 (2009).
Erb, A., Krauth, C., Luzhetskyy, A. & Bechthold, A. Differences in the substrate specificity of glycosyltransferases involved in landomycins A and E biosynthesis. Appl. Microbiol. Biotechnol. 83, 1067–1076 (2009).
Rodriguez, L., Rodríguez, D., Olano, C., Braña, A. F., Méndez, C. & Salas, J. A. Functional analysis of OleY L-oleandrosyl 3-O-methyltransferase of the oleandomycin biosynthetic pathway in Streptomyces antibioticus. J. Bacteriol. 183, 5358–5363 (2001).
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A Practical Streptomyces Genetics, (The John Innes Foundation Centre, Norwich, UK, 2000).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning A Laboratory Manual 2nd edn. (Cold Spring Harbor Laboratory, NY, 1989).
Ishikawa, J. & Hotta, K. FramePlot: a new implementation of the frame analysis for predicting protein-coding regions in bacterial DNA with a high G + C content. FEMS Microbiol. Lett. 174, 251–253 (1999).
Cole, P. A. Chaperone-assisted protein expression. Structure 4 239–242 (1996).
Minami, A. & Eguchi, T. Substrate flexibility of vicenisaminyltransferase VinC involved in the biosynthesis of vicenistatin. J. Am. Chem. Soc. 129, 5102–5107 (2007).
Acknowledgements
We thank Professor Craig A, Townsend and Jason W Labonte at the Johns Hopkins University, who helped us with the preparation of this manuscript. This work was supported in part by the Uehara Memorial Foundation (TE), Naito Foundation (TE and FK) and a Grant-in-Aid for Scientific Research from MEXT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to the late Dr C Richard Hutchinson for his exceptional contributions to natural product biosynthesis, engineering and drug discovery.
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kudo, F., Yonezawa, T., Komatsubara, A. et al. Cloning of the biosynthetic gene cluster for naphthoxanthene antibiotic FD-594 from Streptomyces sp. TA-0256. J Antibiot 64, 123–132 (2011). https://doi.org/10.1038/ja.2010.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.145
Keywords
This article is cited by
-
Cervinomycins C1-4 with cytotoxic and antibacterial activity from Streptomyces sp. CPCC 204980
The Journal of Antibiotics (2020)
-
Genome mining of the sordarin biosynthetic gene cluster from Sordaria araneosa Cain ATCC 36386: characterization of cycloaraneosene synthase and GDP-6-deoxyaltrose transferase
The Journal of Antibiotics (2016)
-
Enhancement of FK506 production by engineering secondary pathways of Streptomyces tsukubaensis and exogenous feeding strategies
Journal of Industrial Microbiology and Biotechnology (2013)
-
Genetic transformation of Diaporthe phaseolorum, an endophytic fungus found in mangrove forests, mediated by Agrobacterium tumefaciens
Current Genetics (2012)
