
Abstract
Early blockade of the renin–angiotensin system is successful in delaying the development of hypertension in spontaneously hypertensive rats (SHRs) and ameliorating organ damage by inhibition of the inflammatory response. In this study, we investigated the role of the angiotensin II type 1 receptor (AT1R) in the early renal inflammatory response in SHR. Blood pressure development and renal inflammatory markers were measured in 4-, 8- and 12-week-old SHR and age-matched Wistar Kyoto (WKY) rats. Separate groups of SHRs were transiently treated with the AT1R blocker losartan between 4 and 8 weeks of age. Urinary excretion of the renal injury markers osteopontin and neutrophil gelatinase-associated lipocalin increased in young SHR. Further, renal expression of inflammatory genes was also increased in young SHR. Losartan inhibited the increase of these inflammatory markers. In contrast, gene expression of the renal injury marker and T-cell inducer kidney injury molecule-1 (KIM-1) was reduced in 4-week-old SHR when compared with WKY. Similarly, the T-cell marker CD3 was significantly decreased in 4-week-old SHR. These effects were not antagonized by AT1R blockade. This study confirms the presence of an early renal inflammatory response in SHR that can be blocked by AT1R antagonism. In addition, it demonstrates that KIM-1 does not behave as a pure kidney injury marker in young SHR, but may reflect kidney maturation.
Similar content being viewed by others


Introduction
High-normal blood pressure (BP), also referred to as prehypertension, has been recognized as an increased risk for developing cardiovascular events and hypertension.1 The initiation of anti-hypertensive treatment in prehypertensives has shown promising results in delaying and/or preventing further development of high BP.2 Furthermore, clinical and experimental evidence demonstrates long-lasting effects on BP and target organ damage after anti-hypertensive treatment for a brief period.2, 3, 4, 5, 6, 7 Recent studies suggest a role for immune mechanisms in the development and sustainment of high BP8, 9, 10 and in particular renal inflammation.11 The induction of adaptive immune responses in the kidney is mainly angiotensin II type 1 receptor (AT1R)-mediated and can be blocked by specific receptor blockers and not via a renin–angiotensin system (RAS)-independent BP lowering.12, 13
A series of experiments in the spontaneously hypertensive rat (SHR) has provided evidence that transient prehypertensive treatment (TPT) with RAS blockers is effective in delaying BP increase and the development of target organ damage.3, 4, 5, 6 The critical period in which TPT is most effective has been pinpointed between 4 and 8 weeks of age. In SHR, the development of renal damage is important in the pathogenesis of this model.14 It has been demonstrated that mainly structural alterations in the renal microcirculation determine the long-lasting antihypertensive effects of RAS inhibitors following TPT.15, 16 The underlying mechanisms involved in this process are still insufficiently understood.
We hypothesize that at the stage of prehypertension, renal inflammation is already provoked by mechanisms recently proposed by Harrison et al.8 and hereby having a deteriorating effect on the hypertensive phenotype. We propose that renal inflammation is a crucial factor in the early development of hypertension in SHR. For this purpose, we analyzed the development of high BP and renal inflammation in young SHR and compared them with age-matched Wistar Kyoto (WKY) rats as normotensive controls. Furthermore, we hypothesized that the early inflammatory response is AT1R-mediated and hence, a subgroup of SHR was given TPT with an AT1R-blocker.
Despite TPT, the increase in BP is just delayed and not prevented. This suggests that some pathophysiological processes may be already ongoing before BP starts to rise. Therefore, this study also investigated the presence and involvement of renal injury markers, such as osteopontin (OPN) and kidney injury molecule-1 (KIM-1) in young SHR. These markers are also well-known regulators of inflammation.17, 18 In the kidney, KIM-1 is mainly expressed on tubules and on several cell types of the immune system, for example, T helper 1 (Th1) and Th2 cells.17, 19 Besides inflammation, this study gives a hint that a suppression of KIM-1 may be involved in the development of early renal aberrations, which may lead to the development of hypertension. KIM-1, a type I transmembrane glycoprotein, is not only a well-known biomarker for the early detection of renal injury,20 but is also involved in the regulation of inflammatory responses.19 Furthermore, KIM-1 is implicated in renal regeneration of dedifferentiated proximal tubule cells.21 KIM-1 seems to have different controversial roles, being involved in renal injury as well as in healing.22
The aims of this study are to investigate (a) the expression of markers of renal injury and inflammation during the onset of hypertension in SHR and (b) the role of the AT1R in the expression of these markers.
Methods
Animals
Four-week-old male SHR (N=45) and WKY (N=21) rats were obtained from Charles River (Sulzfeld, Germany). SHRs were randomly divided over two groups: (1) control SHR without any treatment (N=29) and (2) SHR with losartan treatment (N=16). All animal experiments were approved by the animal ethics committee of Maastricht University and were performed in accordance with institutional and international guidelines. All groups had free access to water and food ad libitum and were housed under controlled conditions of temperature (21 °C) and light (12-h light–dark cycle).
Experimental protocol
Animals received TPT between 4 and 8 weeks of age. To check for potential differences in albuminuria, rats were put in metabolic cages for 24 h to collect urine samples at 8 weeks of age, that is, upon TPT, and 4 weeks after stopping treatment. At 4, 8 and 12 weeks of age BP was determined intra-arterially in the conscious state via a fluid-filled catheter, which was introduced into the right femoral artery and shifted upward into the aorta 2 days before the measurement. Next, at each time point, renal blood flow was measured with a flow probe (Transonic Systems, VB series 0.5 nm, Ithaca, NY, USA) for the determination of renal vascular resistance. Afterward, rats were killed by exsanguination via the abdominal aorta and organs were weighed and harvested for further analysis.
Treatment
Losartan (MSD, 20 mg kg–1 day–1 in phosphate-buffered saline (PBS); GIBCO, Invitrogen, Carlsbad, CA, USA) was given via subcutaneously osmotic minipumps implanted under isoflurane anesthesia (1–4%) between 4 and 8 weeks of age. The dosage was based on earlier studies by our group.3, 12
Urinary markers of renal injury
The rat kidney injury panel 1 was purchased from mesoscale diagnostics (MSD; K15162C-1, Meso Scale Discovery, Gaithersburg, MD, USA). This assay detects lipocalin-2 (neutrophil gelatinase-associated lipocalin, NGAL), OPN and KIM-1 in a sandwich immunoassay and uses a competitive assay format to detect albumin. All reagents were provided with the MSD kit. Each 96-well plate had four-carbon electrodes in the bottom of each well, each pre-coated with one of the four anti-kidney injury markers antibodies of interest. The assay was performed according to the manufacturer’s protocol.20 Finally, albumin and the renal injury markers were corrected for urinary creatinine, determined on the Beckman/Coulter Synchron LX 20 (Beckman Coulter, Brea, CA, USA).
Gene expression analysis
Total RNA was isolated from the right kidney of each animal as previously described11 and 1000 ng of RNA was synthesized into complementary DNA according to the manufacturer’s protocol (iScript cDNA synthesis kit, Bio-Rad Laboratories, Hercules, CA, USA). Gene expression was analyzed by quantitative PCR on the CFX96 Real-Time PCR detection system using the iQ SYBR-green supermix (Bio-Rad Laboratories). Renin, angiotensin-converting enzyme and AT1R expression were analyzed to investigate the changes in RAS-related genes in the kidney. In addition, KIM-1, OPN, NGAL, nephrin and plasminogen activator inhibitor-1 (PAI-1) were determined for the analysis of renal injury. As inflammation-related genes, intercellular adhesion molecule-1, vascular cell adhesion protein 1, monocyte chemotactic protein-1, nuclear factor kappa B (NFκB), tumor necrosis factor α (TNFα) and interleukin 1β (IL1β) and IL6 were investigated. Finally, the expression of several specific markers for cells involved in the immune system were determined: CD68 as macrophage marker, CD3 as T-cell marker, forkhead box P3 as regulatory T cell (Treg) marker, T-box expressed on T cells as Th1 cell marker and GATA-binding protein 3 as Th2 cell marker. The primers applied are presented in Table 1. Results were normalized to the housekeeping gene cyclophilin A and relative changes in expression levels were subsequently calculated using BIO-RAD CFX-manager software. Gene expression in the 4-week-old SHRs was normalized to 1 to compare relative changes among all groups.
Histochemistry and immunostaining
The right kidney of each animal was excised, rinsed in PBS (Invitrogen) and fixed in 4% formalin for 24 h before embedding in paraffin. Central cross-sections of 4 μm of the whole kidney including medulla and cortex were deparaffinized and stained for CD3 to determine T-cell infiltration or CD68 to determine macrophage influx.
CD68
First, slides were incubated for 30 min in 0.3% peroxide in PBS to block endogenous peroxidase activity. Primary antibody (mouse-α-rat ED-1,23 1:50 in PBS with 0.1% bovine serum albumin) was added and incubated overnight at 4 °C. Hereafter, slides were incubated for 30 min with secondary antibody (rabbit-α-mouse-biotine, 1:400 in PBS, Sigma, St Louis, MO, USA). Subsequently, AB complex and diaminobenzidine (Dako Denmark A/S, Glostrup, Denmark) was added to the slides to obtain staining of the macrophages. Finally, the tissues were counterstained with hematoxylin and rehydrated in increasing concentrations of ethanol. Between the different steps, slides were washed in PBS.
CD3
First, slides were incubated for 30 min in 0.3% peroxide in PBS to block endogenous peroxidase activity. Next, antigen retrieval was performed by boiling the slides in a Tris EDTA buffer (1.2 g l–1 Tris and 0.37 g l–1 EDTA, pH 9) for 10 min. Primary antibody (mouse-α-human CD3 (DAKO), 1:200 in PBS with 1% bovine serum albumin) was incubated overnight at 4 °C. Hereafter, slides were incubated for 30 min with secondary antibody (rabbit-α-mouse peroxydase conjugated, 1:1000 in PBS, DAKO). Subsequently, Envision anti-rabbit incubation for 30 min and diaminobenzidine (Dako Denmark A/S) was used to obtain staining of the T cells. Finally, the tissues were counterstained with hematoxylin and dehydrated in increasing concentrations of ethanol.
Analysis of CD68 and CD3 was done by light microscopy at a magnification of × 200 by means of 10 images per subject throughout the entire cortex. Positive cells were manually counted and averaged per subject.
Statistical analysis
Data are expressed as mean±s.e.m. and were analyzed by unpaired Student’s t-test, one-way or two-way analysis of variance with post hoc Bonferroni correction where appropriate. Outliers were detected and removed from the data set with the Grubbs outliers test. Differences were considered statistically significant if values<0.05 were reached (*P<0.05, **P<0.01 and ***P<0.001).
Results
Data on body weight, organ weight and number of animals used, are summarized in Table 2 per group, at 4, 8 and 12 weeks of age.
Hemodynamics and renal function
SHR start to develop hypertension from 4 weeks on when mean arterial pressure (MAP) is still at the same level as the normotensive WKY. MAP was significantly increased at 8 weeks of age when compared with age-matched WKY and it increases to approximately 180 mm Hg by the age of 12 weeks. TPT between 4 and 8 weeks with an AT1R blocker completely restored BP to control values. Four weeks after stopping the RAS blockade MAP increased. However, it stayed significantly lower when compared with the age-matched SHR (Figure 1a). Despite an elevation in MAP, renal vascular resistance demonstrated no significant differences when comparing untreated SHR with age-matched WKY at 8 and 12 weeks of age. TPT in SHR inflicted a significant decrease in renal vascular resistance at 8 weeks of age, which disappeared 4 weeks later (Figure 1b). The levels of albumin excretion in the young animals were, as expected, very low. At 8 weeks of age, no significant difference in albuminuria was detected between WKY and SHR. Four weeks later, at the age of 12 weeks, the albumin excretion was increased in the SHR, which resulted in a significantly higher albuminuria when compared with age-matched WKY. In WKY rats, the excretion of albumin decreased from 8 to 12 weeks, whereas in SHR the levels of albumin excretion started to increase. TPT had little to no effect on albuminuria (Table 3).

Blood pressure (a) and renal vascular resistance (b). KW, kidney weight; MAP, mean arterial pressure; RVR, renal vascular resistance; SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (**P-value<0.01, ***P-value<0.001).
Expression of renal RAS components
WKY rats demonstrated higher levels of renal gene expression for renin, angiotensin-converting enzyme and AT1R when compared with SHR almost at all ages. In SHR, losartan treatment led to a highly significant increase in local renin expression at 8 weeks of age, but this increase completely disappeared 4 weeks later. Finally, TPT caused an increase in AT1R expression to a similar level as the WKY (Figures 2a–c).

(a–c) Renal expression of renin–angiotensin system components. ACE, angiotensin-converting enzyme; AT1R, angiotensin II type 1 receptor; SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (*P-value<0.05, ***P-value<0.001).
Renal injury
Gene expression of the T-cell inducer and renal injury marker KIM-1 was already increased at 4 weeks of age in WKY when compared with SHR. In both rats, the renal expression of KIM-1 increases but remained significantly higher in the WKY. TPT in the SHR had no effect on the expression of this marker (Figure 3a). These changes did not reflect in the excretion of KIM-1 protein in urine (Figure 3b). Further, the renal gene expression of OPN and NGAL, both involved in immunity and known markers of renal injury, were unaltered over time (Figures 3c and e). These results are in sharp contrast with the excretion data where OPN was significantly higher in the 12-week-old SHR when compared with the age-matched WKY. Next, NGAL protein excretion was already increased in the SHR at 8 weeks of age and was still significantly higher 4 weeks later. For both OPN and NGAL, TPT had no direct effect at 8 weeks, but seemed to affect the excretion of these markers in 12-week-old SHR to similar levels as observed for WKY (Figures 3d and f). Nephrin expression in the kidney was increased in SHR when compared with WKY at 4 and 8 weeks of age. Whereas losartan treatment in SHR did not affect nephrin gene expression at 8 weeks, 4 weeks later the expression was higher when compared with the untreated SHR (Figure 3g). Finally, PAI-1 expression was unaltered (Figure 3h).

Gene expression (a,c,e,g,h) and urinary excretion (b,d,f) of renal injury markers. KIM-1 kidney injury molecule-1; NGAL, neutrophil gelatinase-associated lipocalin; OPN, osteopontin; PAI-1, plasminogen activator inhibitor-1; SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (*P-value<0.05, **P-value<0.01, ***P-value<0.001).
Renal inflammation
Figure 4 presents the expression of several inflammation-related genes determined in the kidney. At 4 weeks of age, when BP was still normal almost no differences were detected between WKY and SHR. Only tumor necrosis factor α expression was significantly lower in SHR than WKY at that age. A different pattern was observed at 8 weeks of age. All inflammatory genes were increased in SHR when compared with the age-matched WKY. AT1R-blockade for 4 weeks caused normalization of all markers to similar levels as observed in WKY. Surprisingly, at 12 weeks of age all differences in gene expression between SHR and WKY were normalized. Renal gene expression of CD68, a macrophage marker, was significantly increased in WKY at 4 weeks of age (Figure 5a). CD68 expression increased over time in SHR and the difference with age-matched WKY disappeared from 8 weeks on. These results were confirmed on immunohistochemistry (Figure 6). AT1R-blockade caused a minor, not significant decrease, which was still present at 12 weeks of age.

(a–g) Renal expression of inflammation-related genes. ICAM-1, intercellular adhesion molecule-1; IL, interleukin; MCP1, monocyte chemotactic protein-1; NFκB, nuclear factor kappa B; TNFα, tumor necrosis factor α; VCAM-1, vascular cell adhesion protein-1. SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (*P-value<0.05, **P-value<0.01).

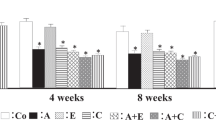
(a–e) Renal gene expression of immune cell markers. FoxP3, forkhead box P3; GATA3, GATA-binding protein 3; ND, not detectable; T-bet, T-box expressed on T cells; SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (*P-value<0.05, **P-value<0.01, ***P-value<0.001).

Immunohistochemical analyses of the macrophage marker CD68. Arrows indicate CD68-positive cells. SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (**P-value<0.01).
Adaptive immune response
Similar to CD68, the general T-cell marker CD3 was significantly increased in WKY when compared with SHR at 4 weeks of age. In contrast, this increase remained present even until the age of 12 weeks. Strikingly, 4 weeks of losartan treatment in SHR caused an increase in renal CD3 expression when compared with untreated SHR, yet, this difference disappeared at 12 weeks (Figure 5b). Analysis by immunohistochemistry confirmed the higher level of CD3-positive cells in young WKY (Figure 7). Next, renal gene expression of T-cell sub-populations demonstrated little differences in Th1 cells (T-box expressed on T cells) and Th2 cells (GATA-binding protein 3; Figures 5d and e). Further, Tregs (forkhead box P3) were undetectable in the kidneys of WKY at 4 weeks of age, whereas they were present in age-matched SHR. Four weeks later, forkhead box P3 expression became apparent in WKY but stayed significantly lower when compared with SHR. At 12 weeks of age, no differences were present anymore (Figure 5c). Four weeks of losartan treatment had no effect on the expression of the different T-cell markers. Only a small increase in T-box expressed on T cells was detected 4 weeks after stopping the AT1R-blockade, that is, at 12 weeks of age.

Immunohistochemical analyses of the T-cell marker CD3. Arrows indicate CD3-positive cells. SHR, spontaneous hypertensive rat; WKY, Wistar Kyoto rat (**P-value<0.01).
Discussion
This study provides novel insights in the contribution of renal inflammation in the early development of hypertension in SHR. First of all, a distinguished increase in inflammatory markers in the kidney was demonstrated at 8 weeks of age in SHR. This was prevented by administration of an AT1R blocker. Second, a suppression of renal KIM-1 expression was found in SHR in the early phase of hypertension development. Finally, the presence of macrophages and T cells in the kidney were surprisingly lower in SHR when compared with WKY. Little to no differences were found in expression markers of the various T-cell sub-populations. Yet, the presence of Treg expression was initially absent in WKY and remained decreased until 8 weeks of age when compared with SHR.
The development of high BP in SHR starts between 4 and 8 weeks of age and reaches a MAP of approximately 175 mm Hg by the age of 12 weeks.3, 24 Hence, the period between 4 and 8 weeks is crucial and underlying processes, such as changes in renal microcirculation, sodium reabsorption and innervations, have been proposed as important contributors to the BP phenotype in SHR.14, 25 Some groups even propose that vascular structure and renal function are already affected before the age of 4 weeks.26 With respect to the strategy of preventing hypertension, transient anti-hypertensive treatments have been given at various ages in the SHR model. Studies with RAS inhibitors, administered in the period from birth until 4 weeks have been unsuccessful and demonstrated even detrimental effects for the littermates,27 whereas perinatal treatment with an NFκB inhibitor had long-term anti-hypertensive effects.28 On the other hand, TPT with RAS blockers between 4 and 8 weeks of age delays the BP increase in SHR,3, 4, 5, 16 as also demonstrated in this study. These studies suggested that RAS blockers antagonize an early immune response that is the starting point for a renal inflammatory process.
The importance of inflammation in the pathogenesis of hypertension is clear, is much debated and subjected to intense research. In SHR, inflammation has been detected in multiple organs, including the heart, vessels and kidney. As BP rises, inflammatory processes become more eminent. However, the presence of early inflammatory processes may be of higher importance by introducing irreversible processes that can lead toward target organ damage. NFκB-positive cells have already been detected at the age of 3 weeks.29 The induction of inflammatory processes can be due to pro-inflammatory mediators, such as Ang II,9 but also directly or indirectly by an increase in BP itself. Another inducer of inflammation at that age might be advanced glycation end products. Despite the absence of oxidative stress, an increase of advanced glycation end product was present in the kidney of SHR when compared with age-matched WKY.30 Whatever the inducer might be, at 8 weeks of age, a clear pattern of increased inflammatory markers, for example, intercellular adhesion molecule-1, vascular cell adhesion protein, monocyte chemotactic protein-1, NFκB, tumor necrosis factor α, IL1β and IL6, was present in the kidney. Despite a BP increase of approximately 35 mm Hg between 8 and 12 weeks, the changes in inflammatory mediators disappeared. Hence, it seems unlikely that the inflammatory mediators are mainly induced by the increase in BP. In addition, at the age of 8 weeks, TPT with an ARB resulted in normalization of all markers. These observations make it plausible that the main induction of renal inflammation at that age happens via AT1R activation, as seen in other models of hypertension.12
According to a recent hypothesis by Harrison et al.8, early inducers of adaptive immunity could be the initiating cause of high BP development. An unexpected result was observed for renal gene expression of CD3, the general T-cell marker. CD3 was upregulated in WKY when compared with SHR. This was confirmed by immunohistochemistry and is in contrast with literature, which demonstrates a higher infiltration of T cells in the kidneys of SHR.29 The same was true for CD68, a macrophage marker. A possible explanation for this can be found in the presence of KIM-1, which besides its function in renal regeneration, acts as a chemo attractant for immune cells.19 Since at young age, KIM-1 is more highly expressed in WKY when compared with SHR, immune cells are more prominently present in the kidneys of these animals. Molecular analysis of markers for the different Th-cell sub-populations, Th1 and Th2, revealed little to no differences over time. Interestingly, Treg expression was present already in young SHR, whereas in young WKY the expression was absent at 4 weeks and remained lower at 8 weeks of age. Since Tregs are involved in the suppression of inflammatory responses,31 this may provide additional evidence for the presence of renal inflammation at early age in SHR.
KIM-1 excretion data suggest that there is little to no renal injury present in young SHR. Hypertensive kidney damage is not morphologically evident before 30 weeks of age.14 This was in line with the absence of albuminuria at 8 weeks of age. Yet, the development of albuminuria precedes the apparent renal pathology and is already slightly increased in SHR 4 weeks later. This was accompanied by a relatively higher excretion of NGAL and OPN, which are both known biomarkers for the early detection of renal disease.20 When minor signs of renal dysfunction come to the surface at 12 weeks of age, it becomes apparent that the increase of albuminuria as well as renal injury markers is delayed and/or prevented by TPT. For albuminuria in SHR, it has already been shown that losartan treatment between 4 and 8 weeks of age resulted in a life-long improvement of renal function until 72 weeks of age, that is, 64 weeks after stopping TPT.3
Whereas no differences were observed in excretion of KIM-1 protein, gene expression data gave some interesting results. From our data it seems that renal KIM-1 expression is highly depressed in SHR when compared with age-matched WKY. As KIM-1 has shown to be involved in renal regeneration after sublethal injury,32 we believe that it might also be important in kidney maturation. In the first 4 weeks after birth, renal development is still ongoing in rats and KIM-1 may be an important regulator during this process. Therefore, the depressed status of KIM-1 observed in SHR might indicate a malfunction in the early renal development causing pathological changes, which result in the development of high BP later on. Of course this conclusion is still speculative and needs further experimentation to unravel underlying mediators and mechanisms. A possible mechanism by which KIM-1 acts might be via its natural ligand tissue inhibiting molecule-4 and hereby the regulation of several inflammatory processes, such as T-cell proliferation.33
As seen in this and many other studies, transient anti-hypertensive treatments can have long-term effects on BP increase and target organ damage. As the focus of this study was on early inflammatory markers in the kidney, the follow-up period was set to only 1-month after TPT. However, previous studies by Baumann et al.3, 4, 15, 16, with a similar experimental setup as this study, provide data on long-term TPT effectiveness in SHR. They demonstrate that 4 weeks of RAS blockade is able to delay the BP increase in SHR. BP is significantly lower until 36 weeks of age, that is, 28 weeks after stopping TPT, and reaches a similar BP level as observed in untreated SHR by the age of 50 weeks.3 Despite the return in BP, effects of TPT on target organs remains visible even until 72 weeks of age. At that advanced age, SHR with TPT have a better cardiac function and less fibrosis and hypertrophy when compared with untreated SHR. In addition, the development of albuminuria is significantly blunted by TPT.3 However, the fact that BP increases although transient anti-hypertensive treatment was given suggest that underlying processes, such as inflammation, are already ongoing before the initiation of treatment. This is supported by the fact that in SHR NFκB seems to be important for the development of hypertension already at birth28 and that vascular remodeling and renal dysfunction are present far before BP starts to increase.14 According to our data this coincides with a depression of renal KIM-1 expression. Altogether, these observations indicate that concerning the initiation of hypertension, different processes are involved at different stages. Proper pinpointing of these processes and combining different treatments at set time points, may provide an appropriate strategy for absolute prevention of the induction of early renal aberrations and consequently stop the development of high BP in later life.
In conclusion, this study provides new insights in the involvement of inflammation during the development of high BP in SHR, a worldwide used model of hypertension. At young age, when BP starts to rise, a peak of renal inflammatory markers is observed, which seems mainly induced by AT1R activation. Second, interesting results were observed regarding KIM-1, a protein involved in inflammation, but also in renal regeneration. A depressed KIM-1 expression in the kidneys of young SHR may already indicate a malfunction in the ongoing renal maturation process. This might be an important contributor in renal structural aberrations long before the hypertensive phenotype becomes apparent in SHR. Further research is needed to strengthen this hypothesis.
References
Vasan RS, Larson MG, Leip EP, Evans JC, O'Donnell CJ, Kannel WB, Levy D . Impact of high-normal blood pressure on the risk of cardiovascular disease. N Engl J Med 2001; 345: 1291–1297.
Julius S, Nesbitt SD, Egan BM, Weber MA, Michelson EL, Kaciroti N, Black HR, Grimm RH Jr., Messerli FH, Oparil S, Schork MA . Feasibility of treating prehypertension with an angiotensin-receptor blocker. N Engl J Med 2006; 354: 1685–1697.
Baumann M, Janssen BJ, Hermans JJ, Peutz-Kootstra C, Witzke O, Smits JF, Struijker Boudier HA . Transient AT1 receptor-inhibition in prehypertensive spontaneously hypertensive rats results in maintained cardiac protection until advanced age. J Hypertens 2007; 25: 207–215.
Baumann M, Megens R, Bartholome R, Dolff S, van Zandvoort MA, Smits JF, Struijker-Boudier HA, De Mey JG . Prehypertensive renin-angiotensin-aldosterone system blockade in spontaneously hypertensive rats ameliorates the loss of long-term vascular function. Hypertens Res 2007; 30: 853–861.
Bergstrom G, Johansson I, Wickman A, Gan L, Thorup C . Brief losartan treatment in young spontaneously hypertensive rats abates long-term blood pressure elevation by effects on renal vascular structure. J Hypertens 2002; 20: 1413–1421.
Harrap SB, Mirakian C, Datodi SR, Lever AF . Blood pressure and lifespan following brief ACE inhibitor treatment in young spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 1994; 21: 125–127.
Luders S, Schrader J, Berger J, Unger T, Zidek W, Bohm M, Middeke M, Motz W, Lubcke C, Gansz A, Brokamp L, Schmieder RE, Trenkwalder P, Haller H, Dominiak P . The PHARAO study: prevention of hypertension with the angiotensin-converting enzyme inhibitor ramipril in patients with high-normal blood pressure: a prospective, randomized, controlled prevention trial of the German Hypertension League. J Hypertens 2008; 26: 1487–1496.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM . Inflammation, immunity, and hypertension. Hypertension 2011; 57: 132–140.
Ruiz-Ortega M, Ruperez M, Esteban V, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J . Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant 2006; 21: 16–20.
Schiffrin EL . Vascular remodeling in hypertension: mechanisms and treatment. Hypertension 2012; 59: 367–374.
Heijnen BFJ, Peutz-Kootstra CJ, Mullins JJ, Janssen BJA, Struijker-Boudier HAJ . Transient renin-angiotensin system stimulation in an early stage of life causes sustained hypertension in rats. J Hypertens 2011; 29: 2369–2380.
Heijnen BF, Nelissen J, van Essen H, Fazzi GE, Cohen Tervaert JW, Peutz-Kootstra CJ, Mullins JJ, Schalkwijk CG, Janssen BJ, Struijker-Boudier HA . Irreversible renal damage after transient ras stimulation: involvement of an at1-receptor mediated immune response. PLoS One 2013; 8: e57815.
Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, Fujita T . Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension 2003; 42: 31–38.
Hultstrom M . Development of structural kidney damage in spontaneously hypertensive rats. J Hypertens 2012; 30: 1087–1091.
Baumann M, Bartholome R, Peutz-Kootstra CJ, Smits JF, Struijker-Boudier HA . Sustained tubulo-interstitial protection in SHRs by transient losartan treatment: an effect of decelerated aging? Am J Hypertens 2008; 21: 177–182.
Baumann M, Janssen BJ, Rob Hermans JJ, Bartholome R, Smits JF, Struijker Boudier HA . Renal medullary effects of transient prehypertensive treatment in young spontaneously hypertensive rats. Acta Physiol (Oxf) 2009; 196: 231–237.
Nozaki Y, Nikolic-Paterson DJ, Snelgrove SL, Akiba H, Yagita H, Holdsworth SR, Kitching AR . Endogenous Tim-1 (Kim-1) promotes T-cell responses and cell-mediated injury in experimental crescentic glomerulonephritis. Kidney Int 2012; 81: 844–855.
Wolak T, Kim H, Ren Y, Kim J, Vaziri ND, Nicholas SB . Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int 2009; 76: 32–43.
Umetsu SE, Lee WL, McIntire JJ, Downey L, Sanjanwala B, Akbari O, Berry GJ, Nagumo H, Freeman GJ, Umetsu DT, DeKruyff RH . TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol 2005; 6: 447–454.
Devarajan P . The use of targeted biomarkers for chronic kidney disease. Adv Chronic Kidney Dis 2010; 17: 469–479.
Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV . Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem 2002; 277: 39739–39748.
Lim AI, Tang SC, Lai KN, Leung JC . Kidney injury molecule-1: more than just an injury marker of tubular epithelial cells? J Cell Physiol 2012; 228: 917–924.
Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD . Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 1994; 83: 140–147.
Reckelhoff JF, Zhang H, Granger JP . Decline in renal hemodynamic function in aging SHR: role of androgens. Hypertension 1997; 30 (3 Pt 2): 677–681.
Gattone VH 2nd, Evan AP, Overhage JM, Severs WB . Developing renal innervation in the spontaneously hypertensive rat: evidence for a role of the sympathetic nervous system in renal damage. J Hypertens 1990; 8: 423–428.
Komolova M, Friberg P, Adams MA . Altered vascular resistance properties and acute pressure-natriuresis mechanism in neonatal and weaning spontaneously hypertensive rats. Hypertension 2012; 59: 979–984.
Racasan S, Hahnel B, van der Giezen DM, Blezer EL, Goldschmeding R, Braam B, Kriz W, Koomans HA, Joles JA . Temporary losartan or captopril in young SHR induces malignant hypertension despite initial normotension. Kidney Int 2004; 65: 575–581.
Koeners MP, Braam B, Joles JA . Perinatal inhibition of NF-kappaB has long-term antihypertensive effects in spontaneously hypertensive rats. J Hypertens 2011; 29: 1160–1166.
Rodriguez-Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND . Evolution of renal interstitial inflammation and NF-kappaB activation in spontaneously hypertensive rats. Am J Nephrol 2004; 24: 587–594.
Baumann M, Stehouwer C, Scheijen J, Heemann U, Struijker Boudier H, Schalkwijk C . N epsilon-(carboxymethyl)lysine during the early development of hypertension. Ann N Y Acad Sci 2008; 1126: 201–204.
Murphy TJ, Ni Choileain N, Zang Y, Mannick JA, Lederer JA . CD4+CD25+ regulatory T cells control innate immune reactivity after injury. J Immunol 2005; 174: 2957–2963.
Yamanishi Y, Kitaura J, Izawa K, Kaitani A, Komeno Y, Nakamura M, Yamazaki S, Enomoto Y, Oki T, Akiba H, Abe T, Komori T, Morikawa Y, Kiyonari H, Takai T, Okumura K, Kitamura T . TIM1 is an endogenous ligand for LMIR5/CD300b: LMIR5 deficiency ameliorates mouse kidney ischemia/reperfusion injury. J Exp Med 2010; 207: 1501–1511.
Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE, Kenny J, Zheng XX, Umetsu DT, DeKruyff RH, Strom TB, Kuchroo VK . TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol 2005; 6: 455–464.
Acknowledgements
We thank Gregorio Fazzi for expert technical assistance. This work was performed within the framework of Dutch Top Institute Pharma, project ‘Renin-Angiotensin system blockade beyond Angiotensin II (T2-301)’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Heijnen, B., Van Essen, H., Schalkwijk, C. et al. Renal inflammatory markers during the onset of hypertension in spontaneously hypertensive rats. Hypertens Res 37, 100–109 (2014). https://doi.org/10.1038/hr.2013.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2013.99
Keywords
This article is cited by
-
RNA Sequencing Reveals Novel Transcripts from Sympathetic Stellate Ganglia During Cardiac Sympathetic Hyperactivity
Scientific Reports (2018)
-
Phthalate exposure as a risk factor for hypertension
Environmental Science and Pollution Research (2018)
-
Early urinary biomarkers for renal tubular damage in spontaneously hypertensive rats on a high salt intake
Hypertension Research (2016)
-
Elevated circulating sST2 associated with subclinical atherosclerosis in newly diagnosed primary hypertension
Hypertension Research (2016)
-
Increased neutrophil to lymphocyte ratio in persons suffering from hypertension with hyperhomocysteinemia
Hypertension Research (2016)
