
Abstract
A Japanese family with autosomal recessive cerebellar ataxia type 8 (SCAR8, MIM 610743) is described. We identified a novel SYNE1 frameshift deletion (c.6843del, p.Q2282Sfs*3). This family shared similar clinical manifestations characterized by adult-onset, relatively pure cerebellar ataxia with mild eye movement abnormality. Intelligence and bulbar and respiratory functions were unaffected. This study suggests the clinical utility of using panel-based exome sequencing for genetic diagnosis in hereditary ataxias in a cost-efficient manner.
Similar content being viewed by others


Hereditary spinocerebellar ataxias (SCAs) are a group of genetically and clinically heterogeneous disorders. Autosomal recessive cerebellar ataxia type 8 (SCAR8, also referred to as autosomal recessive cerebellar ataxia type 1 (ARCA1) or recessive ataxia of Beauce) (MIM 610743) is one of the autosomal recessive spinocerebellar ataxias (SCARs) and was initially described in 53 French–Canadian families.1 Affected members of French–Canadian families are clinically characterized by late-onset, slow progression of relatively pure cerebellar ataxia.1,2 The disease-causing gene (SYNE1) encodes Nesprin-1 (nuclear envelope spectrin 1), an exceptionally large protein composed of 8,749 amino acids and two calponin homology domains, followed by 74 spectrin repeats and a C-terminal Klarsicht/ANC-1/Syne homology domain (KASH). Nesprin-1 is a member of the spectrin family and plays an essential role in nuclear-cytoskeletal connections.3
Following recent progress in the development of high-throughput sequencing technologies, SCAR8-causing variants in SYNE1 have been reported worldwide.4–14 Moreover, the clinical presentations of SCAR8 have expanded with variable combinations of cerebellar and extra-cerebellar neurological dysfunctions, including amyotrophic lateral sclerosis-like motor neuron features.4,9–11 However, little is known about how the loss of Nesprin-1 function causes the unique phenotypic variation of SCAR8.
Here, we report a Japanese SCAR8 family carrying a novel homozygous frameshift deletion in SYNE1 based on panel-based targeted exome sequencing. To the best of our knowledge, this is the fourth SCAR8 family reported in Japan.4 We determined that they were carrying a novel homozygous frameshift deletion (c.6843delT, p.Q2282Sfs*3) in SYNE1 using panel-based targeted exome sequencing.
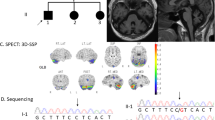
The proband (patient 1, IV-5), a 67-year-old Japanese woman, was born to consanguineous parents (Figure 1a). She had a family history of gait difficulty and dysarthria in her elder brother (IV-1), mother (III-12), and maternal grandmother (II-4). At age 22, she first noticed that her speech appeared to be slurred. She began falling while walking at age 35. A neurological examination at age 67 showed horizontal gaze-evoked nystagmus and saccadic intrusions during smooth pursuits. Her speech was slurred, but dysphagia was absent. She showed limb and truncal ataxia. There was no evidence of pyramidal sign, muscle atrophy, or sensory disturbances. Her total scale for assessment and rating of ataxia score was 26, and her inventory of non-ataxia signs score was 2. Her mini-mental state examination score was 24. Brain magnetic resonance imaging (MRI) at age 65 revealed atrophy in the cerebellar hemispheres and vermis. Cortical or brainstem atrophy was not evident (Figure 1b, c).

Clinical features of a family with a novel SYNE1 mutation. (a) Pedigree of the family. (b–e) T1-weighted brain MRI showed marked atrophy in the cerebellar hemispheres and vermis. b, c Patient 1. d, e Patient 2. ■/●, affected individuals; /, deceased; →, proband; MRI, magnetic resonance imaging.
Patient 2 (IV-1), a 74-year-old man, was the elder brother of patient 1. He suffered from gait difficulty at approximately age 30 and dysarthria at age 35. He became unable to walk at age 65. His neurological examination at age 74 showed saccadic eye movement, dysarthria, ataxic speech, limb, and truncal ataxia. There was no evidence of bulbar palsy, pyramidal sign, muscle atrophy, or sensory disturbances. His scale for assessment and rating of ataxia score was 35, inventory of non-ataxia signs score was 2, and mini-mental state examination score was 23. Brain MRI at age 73 showed atrophy in the cerebellar hemispheres and vermis (Figure 1d, e).
Patient 3 (III-12) was the mother of patients 1 and 2. She was also born to consanguineous parents and showed cerebellar ataxia. She first noticed gait difficulty and slurred speech at approximately age 30 and became unable to walk at age 40. She died at age 91.
Molecular diagnosis was performed on genomic DNA extracted from a blood sample after informed consent was obtained from patients 1 and 2 and their unaffected sister (IV-3). First, screening for common inherited dominant repeat expansion SCA genes, i.e., SCA1, SCA2, MJD/SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, SCA31, and DRPLA was negative for patient 1. Next, to screen multiple SCA genes and other known ataxia-associated genes, we performed panel-based targeted exome sequencing for patient 1 using a TruSight One Sequencing Panel (Illumina, San Diego, CA, USA) and a MiSeq benchtop sequencer (Illumina). For data analysis, we used VariantStudio, version 2.2.1 (Illumina). This study was approved by the ethics committee at Shinshu University.
We found a homozygous novel deletion NM_033071.3(SYNE1_v001):c.6843del in exon 46 of SYNE1 for patient 1. This deletion results in a frameshift at amino acid 2,285 out of 8,749, truncating the encoded protein (p.Q2282Sfs*3). No other mutations were detected in the coding regions of other ataxia-related genes (data not shown). This result was confirmed using Sanger sequencing, whereas the patient’s affected brother (IV-1) was homozygous and her unaffected sister (IV-3) was heterozygous for this mutation (Figure 2a). This mutation has not been described in human genome variation databases, including the 1000 Genomes Project (http://www.internationalgenome.org), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org) and Human Genetic Variation Database (HGVD) (http://www.genome.med.kyoto-u.ac.jp/SnpDB/), or in disease-causing mutation databases, such as the Human Gene Mutation Database (HGMD) Professional 2017.8 (http://www.hgmd.org/) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/).

Mutation in SYNE1 observed in the present family. (a) Direct nucleotide sequencing of PCR-amplified DNA around codon 6,843 on exon 46 of the SYNE1 gene. The arrow head denotes the deleted base. The DNA and corresponding amino-acid sequences of the wild-type and mutant SYNE1 alleles are also shown. (b) Schematic diagram of Nesprin-1 and some of the major alternating isoforms, and locations of reported SYNE1 variants. The location of the mutation in this study is denoted by the arrow head. Circle variants are missense variants, triangles are splice site variants, and rectangles are truncated variants. Biallelic variants are shown in a solid color. AMC, autosomal recessive arthrogryposis multiplex congenital; CH, a pair of N-terminal CH domains; KASH, C-terminal Klarsicht/ANC-1/Syne homology (KASH) domain; MND, motor neuron disease; Others, autism and pontocerebellar hypoplasia; SCAR8, autosomal recessive cerebellar ataxia type 8; SR, spectrin repeats.
The clinical manifestations of the family in this report are characterized by adult-onset, relatively pure cerebellar ataxia with mild eye movement abnormality. The overall clinical courses of our patients were very protracted. Brain images showed marked cerebellar atrophy, whereas the midbrain and pons were spared. These clinical features are similar to those of the aforementioned French–Canadian families.2
Figure 2b shows the protein diagram of Nesprin-1, illustrating the locations of the 97 variants that were registered in HGMD Professional 2017.8 as ‘disease-causing mutations.’ In SCAR8, most of the disease causative variants are distributed across the entire gene, excluding the KASH domain (Figure 2b). Nesprin-1 has multiple alternative start and termination sites throughout SYNE1, allowing the generation of smaller alternating isoforms.3 Razafsky et al.15 noted that full-length Nesprin-1 (Nesprin-1 giant) and one of the isoforms of Nesprin-1 giant devoid of the KASH (KLNes1g) are specifically expressed in the central nervous system, whereas KLNes1g is expressed most abundantly expressed in the cerebellum. The detected mutation in our family could truncate multiple Nesprin-1 isoforms, including Nesprin-1 giant and KLNes1g (Figure 2b).
SYNE1 is also known as the disease-causing gene of autosomal dominant Emery–Dreifuss muscular dystrophy (EDMD4)16 and autosomal recessive arthrogryposis multiplex congenital (AMC).17 Most of the Emery–Dreifuss muscular dystrophy-causative mutations were heterozygous missense, suggesting that the pathogenesis of Emery–Dreifuss muscular dystrophy involves gain-of-function effects on Nesprin-1 (Figure 2b). All arthrogryposis multiplex congenital-causative mutations would be expected to lack most of the KASH-containing isoforms,17,18 such as Nesprin-1α2, which is known as one of the predominantly muscle-specific isoforms.19 In contrast, SCAR8-causative mutations are likely to affect Nesprin-1 giant and KLNes1g, and not affect the short KASH-containing isoforms. The nine SCAR8-causative mutations, which can truncate the Nesprin-1α2 isoform, were compound heterozygous with truncating variants outside the nesprin-1α2 region (Figure 2b). These findings support the hypothesis that loss of the short KASH-containing alternative isoforms is sufficient to cause SYNE1-associated arthrogryposis multiplex congenital.18 In SCAR8, there was no obvious correlation between the position of the truncation variants and clinical phenotype, which presented with and without motor neuron features (Figure 2b). The pathological mechanisms underlying the SYNE1 mutations that cause phenotypic variations of SCAR8 remain to be elucidated.
In Japan, SCARs account for 1.8% of patients with ataxia.20 However, the disease frequency of each subtype in SCARs is unclear because of a high level of genetic heterogeneity or involvement of large genes. Whole-exome sequencing is recognized as an effective tool for the diagnosis of rare diseases, whereas the cost of whole-exome sequencing is still unaffordable for most patients. This study suggests the clinical utility of using panel-based targeted exome sequencing for genetic diagnosis in hereditary ataxias. Studies involving an increased number of patients with SYNE1 mutations are needed to gain further insight into the molecular mechanisms of SYNE1-related diseases.
Additional infomation
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Gros-Louis F, Dupré N, Dion P, Fox MA, Laurent S, Verreault S et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet 2007; 39: 80–85.
Dupré N, Gros-Louis F, Chrestian N, Verreault S, Brunet D, de Verteuil D et al. Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann Neurol 2007; 62: 93–98.
Rajgor D, Shanahan CM . Nesprins: from the nuclear envelope and beyond. Expert Rev Mol Med 2013; 15: e5.
Izumi Y, Miyamoto R, Morino H, Yoshizawa A, Nishinaka K, Udaka F et al. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology 2013; 80: 600–601.
Noreau A, Bourassa CV, Szuto A, Levert A, Dobrzeniecka S, Gauthier J et al. SYNE1 mutations in autosomal recessive cerebellar ataxia. JAMA Neurol 2013; 70: 1296–1231.
Fogel BL, Lee H, Deignan JL, Strom SP, Kantarci S, Wang X et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol 2014; 71: 1237–1246.
Hamza W, Ali Pacha L, Hamadouche T, Muller J, Drouot N, Ferrat F et al. Molecular and clinical study of a cohort of 110 Algerian patients with autosomal recessive ataxia. BMC Med Genet 2015; 16: 36.
Keogh MJ, Steele H, Douroudis K, Pyle A, Duff J, Hussain R et al. Frequency of rare recessive mutations in unexplained late onset cerebellar ataxia. J Neurol 2015; 262: 1822–1827.
Özoğuz A, Uyan Ö, Birdal G, Iskender C, Kartal E, Lahut S et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging 2015; 36: 1764.e9–1764.e18.
Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmüller C, Baets J et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain 2016; 139: 1378–1393.
Mademan I, Harmuth F, Giordano I, Timmann D, Magri S, Deconinck T et al. Multisystemic SYNE1 ataxia: confirming the high frequency and extending the mutational and phenotypic spectrum. Brain 2016; 139: e46.
Gama MT, Houle G, Noreau A, Dionne-Laporte A, Dion PA, Rouleau GA et al. SYNE1 mutations cause autosomal-recessive ataxia with retained reflexes in Brazilian patients. Mov Disord 2016; 31: 1754–1756.
Wiethoff S, Hersheson J, Bettencourt C, Wood NW, Houlden H . Heterogeneity in clinical features and disease severity in ataxia-associated SYNE1 mutations. J Neurol 2016; 263: 1503–1510.
Algahtani H, Marzouk Y, Algahtani R, Salman S, Shirah B . Autosomal recessive cerebellar ataxia type 1 mimicking multiple sclerosis: a report of two siblings with a novel mutation in SYNE1 gene in a Saudi family. J Neurol Sci 2017; 372: 97–100.
Razafsky D, Hodzic D . A variant of nesprin1 giant devoid of kash domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol Dis 2015; 78: 57–67.
Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet 2007; 16: 2816–2833.
Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M et al. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet 2009; 18: 3462–3469.
Baumann M, Steichen-Gersdorf E, Krabichler B, Petersen BS, Weber U, Schmidt WM et al. Homozygous SYNE1 mutation causes congenital onset of muscular weakness with distal arthrogryposis: a genotype-phenotype correlation. Eur J Hum Genet 2017; 25: 262–266.
Zhang Q, Skepper JN, Yang F, Davies JD, Hegyi L, Roberts RG et al. Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J Cell Sci 2001; 114: 4485–4498.
Tsuji S, Onodera O, Goto J, Nishizawa M, Diseases Study Group on Ataxic Diseases. Sporadic ataxias in Japan--a population-based epidemiological study. Cerebellum 2008; 7: 189–197.
Data Citations
Nakamura, Katsuya HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1732 (2017)
Acknowledgements
The authors thank Rie Kawamura, Emiko Kise, Yuki Takahashi, Emi Nomura, and Sonomi Nagasaki for technical support and helpful discussions. This study was supported in part by the Research Committee of Ataxia, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health and Labour Sciences Research Grants, The Ministry of Health, Labour and Welfare, Japan; a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology (Problem-Solving Oriented Training Program for Advanced Medical Personnel: NGSD Project); and a Grant-in-Aid for Initiative on Rare and Undiagnosed Diseases (IRUD) from Japan Agency for Medical Research and Development, AMED.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yoshinaga, T., Nakamura, K., Ishikawa, M. et al. A novel frameshift mutation of SYNE1 in a Japanese family with autosomal recessive cerebellar ataxia type 8. Hum Genome Var 4, 17052 (2017). https://doi.org/10.1038/hgv.2017.52
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.52
This article is cited by
-
Autosomal recessive adult onset ataxia
Journal of Neurology (2022)
-
Autosomal Recessive Cerebellar Ataxia Type 1: Phenotypic and Genetic Correlation in a Cohort of Chinese Patients with SYNE1 Variants
The Cerebellum (2021)
