
Abstract
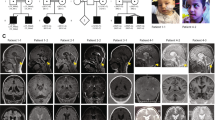
Spinocerebellar ataxia, autosomal recessive-17 (SCAR17) is a rare hereditary ataxia characterized by ataxic gait, cerebellar signs and occasionally accompanied by intellectual disability and seizures. Pathogenic mutations in the CWF19L1 gene that code for CWF19 like cell cycle control factor 1 cause SCAR17. We report here two unrelated families with the clinical characteristics of global developmental delay, cerebellar ataxia, pyramidal signs, and seizures. Cerebellar atrophy, and T2/FLAIR hypointense transverse pontine stripes were observed in brain imaging. Exome sequencing identified novel homozygous mutations including a splice acceptor site variant c.1375-2 A > G on intron 12 in a male patient and a single nucleotide variant c.452 T > G on exon 5 resulting in a missense variant p.Ile151Ser in the female patient from two unrelated families, respectively. Sanger sequencing confirmed the segregation of these variants in the family members with autosomal recessive inheritance. Transcript analysis of the splice site variant revealed activation of a novel cryptic splice acceptor site on exon 13 resulting in an alternative transcription with a loss of nine nucleotides on exon 13. Translation of this transcript predicted an in-frame deletion of three amino acids p.(459_461del). We also observed a novel exon 13 skipping which results in premature termination of the protein product. Our study expands the phenotype, radiological features, and genotypes known in SCAR17.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The data in this study is available with the corresponding author upon reasonable request. The data are not publicly available due to patient privacy and ethical considerations.
References
Jayadev S, Bird TD. Hereditary ataxias: overview. Genet Med. 2013;15:673–83.
Nguyen M, Boesten I, Hellebrekers DM, Vanoevelen J, Kamps R, de Koning B, et al. Pathogenic CWF19L1 variants as a novel cause of autosomal recessive cerebellar ataxia and atrophy. Eur J Hum Genet. 2016;24:619–22.
Burns R, Majczenko K, Xu J, Peng W, Yapici Z, Dowling JJ, et al. Homozygous splice mutation in CWF19L1 in a Turkish family with recessive ataxia syndrome. Neurology 2014;83:2175–82.
Evers C, Kaufmann L, Seitz A, Paramasivam N, Granzow M, Karch S, et al. Exome sequencing reveals a novel CWF19L1 mutation associated with intellectual disability and cerebellar atrophy. Am J Med Genet A 2016;170:1502–9.
Algahtani H, Shirah B, Almatrafi S, Al-Qahtani MH, Abdulkareem AA, Naseer MI. A novel variant in CWF19L1 gene in a family with late-onset autosomal recessive cerebellar Ataxia 17. Neurol Res. 2021;43:141–7.
Shi J, Lv S, Wu M, Wang X, Deng Y, Li Y, et al. HOTAIR-EZH2 inhibitor AC1Q3QWB upregulates CWF19L1 and enhances cell cycle inhibition of CDK4/6 inhibitor palbociclib in glioma. Clin Transl Med. 2020;10:182–98.
Masaki S, Yoshimoto R, Kaida D, Hata A, Satoh T, Ohno M, et al. Identification of the specific interactors of the human lariat RNA debranching enzyme 1 protein. Int J Mol Sci. 2015;16:3705–21.
Ohi MD, Link AJ, Ren L, Jennings JL, McDonald WH, Gould KL. Proteomics analysis reveals stable multiprotein complexes in both fission and budding yeasts containing Myb-related Cdc5p/Cef1p, novel pre-mRNA splicing factors, and snRNAs. Mol Cell Biol. 2002;22:2011–24.
Wang S, Wang H, Liu J, Zhang X, Yang Y, Lu C, et al. Expression patterns and functional analysis of porcine lnc-34015. Anim Biotechnol. 2022:1–11.
Ruan M, Wang H, Zhu M, Sun R, Shi J, Wang Q, et al. Heterozygous pathogenic variants in CWF19L1 in a Chinese family with spinocerebellar ataxia, autosomal recessive 17. J Clin Lab Anal. 2022;36:e24767.
Alvarez C, Grimmel M, Ebrahimi-Fakhari D, Paul VG, Deininger N, Riess A, et al. Expansion of the phenotypic and molecular spectrum of CWF19L1-related disorder. Clin Genet. 2023;103:566–73.
Muthusamy B, Selvan LDN, Nguyen TT, Manoj J, Stawiski EW, Jaiswal BS, et al. Next-generation sequencing reveals novel mutations in X-linked intellectual disability. OMICS 2017;21:295–303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–54.
Ahmad M, Sinha A, Ghosh S, Kumar V, Davila S, Yajnik CS, et al. Inclusion of population-specific reference panel from India to the 1000 Genomes Phase 3 panel improves imputation accuracy. Sci Rep. 2017;7:6733.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Kumari R, Holla VV, Phulpagar P, Sriram N, Hegde AG, Vengalil S, et al. Whole exome sequencing and transcript analysis discover a novel pathogenic splice site mutation in DCAF17 gene underlying Woodhouse-Sakati syndrome. J Neuroendocrinol. 2022;34:e13185.
Pravinbabu P, Holla VV, Phulpagar P, Kamble N, Netravathi M, Yadav R, et al. A splice altering variant in NDRG1 gene causes Charcot-Marie-Tooth disease, type 4D. Neurol Sci. 2022;43:4463–72.
Bandari AK, Muthusamy B, Bhat S, Govindaraj P, Rajagopalan P, Dalvi A, et al. A novel splice site mutation in IFNGR2 in patients with primary immunodeficiency exhibiting susceptibility to mycobacterial diseases. Front Immunol. 2019;10:1964.
Muthusamy B, Nguyen TT, Bandari AK, Basheer S, Selvan LDN, Chandel D, et al. Exome sequencing reveals a novel splice site variant in HUWE1 gene in patients with suspected Say-Meyer syndrome. Eur J Med Genet. 2020;63:103635.
Sandford E, Burmeister M. Genes and genetic testing in hereditary ataxias. Genes (Basel). 2014;5:586–603.
Yapici Z, Eraksoy M. Non-progressive congenital ataxia with cerebellar hypoplasia in three families. Acta Paediatr. 2005;94:248–53.
Acknowledgements
We are grateful to the families for providing informed consent to participate in this study.
Funding
We thank Science and Engineering Research Board (SERB), Department of Science & Technology, Government of India (EMR/2017/004853) for providing funding to carry out a part of this study and for providing research fellowship to Prashant Phulpagar.
Author information
Authors and Affiliations
Contributions
BM and VVH conceived the idea and designed the study. Sample collection, processing, data analysis, and interpretation of the results were performed by BM and PP. Experiments were performed by PP and DT. First draft of the manuscript and Figure illustrations were prepared by PP and VVH. PKP, VVH, RY, and NK performed a clinical assessment of the patient and provided samples. All authors reviewed the manuscript, provided critical comments to improve the draft, and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the ethics committee at the National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore. This study was performed after getting appropriate informed consents from the patient and relatives.
Consent for publication
We obtained informed written consent from patient and his relatives to publish this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Phulpagar, P., Holla, V.V., Tomar, D. et al. Novel CWF19L1 mutations in patients with spinocerebellar ataxia, autosomal recessive 17. J Hum Genet 68, 859–866 (2023). https://doi.org/10.1038/s10038-023-01195-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-023-01195-5
