
Abstract
Purpose:
High myopia is one of the leading causes of blindness worldwide, with high heritability. However, only a few causative genes have been identified, and the pathogenesis is still unclear. Our aim was to identify a novel causative gene in a family with autosomal-dominant, nonsyndromic high myopia.
Methods:
Whole-genome linkage and whole-exome sequencing were conducted on the family. Real-time quantitative polymerase chain reaction and immunoblotting were applied to test the functional consequence of the candidate mutation. Sanger sequencing was performed to screen for the candidate gene in other families or sporadic cases.
Results:
A heterozygous missense mutation, c.871G>A (p.Glu291Lys), within P4HA2 was cosegregating with the phenotype in the family. The segregating mutation caused premature degradation of unstable messenger RNA. Subsequent screening for P4HA2 in 186 cases identified an additional four mutations in 5 cases, including the frameshift mutation c.1349_1350delGT (p.Arg451Glyfs*8), the nonsense mutation c.1327A>G (p.Lys443*), and two deleterious missense mutations, c.419A>G (p.Gln140Arg) and c.448A>G (p.Ile150Val). The missense mutation c.419A>G (p.Gln140Arg) was recurrently identified in a sporadic case and was segregating in a three-generation family.
Conclusion:
P4HA2 was identified as a novel causative gene for nonsyndromic high myopia. This study also indicated that the disruption of posttranslational modifications of collagen is an important factor in the pathogenesis of high myopia.
Genet Med 17 4, 300–306.
Similar content being viewed by others

Introduction
Myopia is the most common ocular abnormality worldwide. The prevalence is 23.8% in Europe1 and 64.9% in China.2 The prevalence of high myopia, which is clinically defined as a spherical equivalent refractive error ≥−6.0 diopter spheres has risen to 2.1% in Europe1 and 4.3% in China.2 High myopia may result in several severe complications, including degenerative changes in the sclera, choroid, and retinal pigment epithelium; retinal detachment; cataract; and glaucoma; and it greatly increases the risk of blindness.3 Uncorrected refractive error is the second most common cause of blindness, accounting for the cause in ~21% of all blind persons.4 High myopia has a large public health impact because of its extremely high prevalence and because it is the leading cause of blindness.
Epidemiology studies demonstrated that high myopia has a very high heritability.5 Genome-wide association studies have identified dozens of genes susceptible to myopia risk, which are involved in neurotransmitter functions (GJD2, RASGRF1, and GRIA4), retinoic acid metabolism (RDH5, RGR, and RORB), and ion channel activity (KCNQ5, KCNJ2, KCNMA1, and CACNA1D) or are involved in ocular and central nervous system development (SIX6, CHD7, ZIC2, and PRSS56).6 For high myopia, common associated variants have been identified in several genes or loci, such as 11q24.1, 13q12.12, 4q25, 1q14, CTNND2, SNTB1, VIPR2, and ZFHX1B.6 However, these genes or loci were poorly replicated, except for SNTB1, which was identified in two independent genome-wide association studies.7,8 Family-based linkage analysis also identified many linkage loci containing potential rare causative variants or genes (OMIM). Using family-based conversional positional cloning or whole-exome sequencing, four genes corresponding to autosomal-dominant high myopia (ZNF644,9 CCDC111,10 SCO2,11 and SLC39A512) and two genes (LEPREL113 and LRPAR114) corresponding to autosome-recessive high myopia were recently identified. These gene mutations resulted in only a small proportion of high myopia, however, and the causative variants or genes located in most of the linkage loci are still unresolved. Because of the limitations of genetic identification, the pathogenesis remains unclear.
In this study, using whole-genome linkage analysis and whole-exome sequencing, we identified a new causative gene, P4HA2, for nonsyndromic high myopia in a Chinese family. Mutation screening for P4HA2 in additional families with high myopia and sporadic cases identified an additional four mutations. Importantly, one of the identified mutations was also segregating in a three-generation family.
Materials and Methods
Subjects and clinical investigation
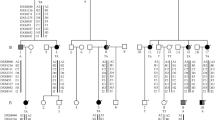
A family with nonsyndromic high myopia (HM-SR3) with autosomal-dominant inheritance ( Figure 1a ) from a Chinese population participated in this study. Thirteen members (nine affected; Figure 1a ) from family HM-SR3 were recruited and underwent a clinical examination and blood collection after providing informed consent. Comprehensive ophthalmic examinations, including visual acuity, refraction, slit-lamp biomicroscopy, indirect ophthalmoscopy, and measurement of axial length via type A ultrasonography, were performed for all of the members, and the refractive error and axial length were measured and recorded. The affected individuals also had a fundus photograph taken, if applicable. We recruited 186 family probands or sporadic cases with high myopia for mutation screening of the candidate gene. This study was approved by the institutional review board of the State Key Laboratory of Medical Genetics.

Variant c.871G>A (p.E291K) within P4HA2 segregated with high myopia phenotypes and caused unstable messenger RNA. (a) The pedigree plot for HM-SR3: solid symbols represent affected individuals; M numbers denote individuals whose DNA samples were analyzed. Individuals highlighted by a red dashed circle were selected for whole-exome sequencing. The missense mutation (E291K) identified by whole-genome linkage and whole-exome sequencing segregated with the phenotype exactly. (b) Multipoint parametric linkage analysis demonstrated four linkage signals with maximum LOD scores of more than 2 on chromosomes 1, 5, 6, and 21. (c) Variant filtering of whole-exome sequencing data–combined linkage results directly identified the segregating mutation. (d) Real-time quantitative polymerase chain reaction revealed that the messenger RNA expression level was significantly decreased in familial cases compared with normal controls. (e) Immunoblot revealed that the protein level of p4ha2 was remarkably decreased in familial cases compared with normal controls.
Whole-genome genotyping and linkage analysis
Genomic DNA was extracted from leukocytes of all recruited family members via the standard proteinase K digestion and phenol-chloroform method. Whole-genome genotyping was performed using the Illumina HumanCytoSNP-12 DNA Analysis BeadChip (Illumina, San Diego, CA) on all recruited members strictly following the instructions of the Illumina protocol. Genotype calling and quality control were performed using the Illumina GenomeStudio Genotyping Module. Single-nucleotide polymorphisms with Mendelian inconsistencies or call rates lower than 100% were excluded. Individual(s) were genotyped again if the call rate was lower than 98%. Mendelian errors were checked using the MERLIN (version 1.12) PEDSTATS program.15 We selected autosomal single-nucleotide polymorphisms with minor allele frequencies >0.3 (n = 47,606) for linkage analyses. The left single-nucleotide polymorphisms were uniformly distributed across the chromosomes. Parametric multipoint linkage analysis using the genotypes after quality control were performed with the MERLIN program based on an autosomal-dominant model with 0.9 penetrance and an allele frequency of 0.01. The affection statuses were strictly defined according to the clinical diagnoses. The detailed whole-genome genotyping method and linkage analysis procedures are described in our previous study.12
Whole-exome sequencing and bioinformatics analysis
For each individual selected for whole-exome sequencing, 1 µg of genomic DNA was fragmented and selected (aiming for a 350- to 400-base pair product) and was amplified with polymerase chain reaction (PCR). Exome capturing was performed to collect the protein-coding regions of human genome DNA using Agilent SureSelect Human All Exon V4+UTRs enrichment platform (Agilent Technologies, Santa Clara, CA), according to the manufacturer’s instructions. The exon-enriched DNA libraries were sequenced using the Illumina HiSeq 2000 platform (Illumina, San Diego, CA), following the manufacturer’s instructions. Generated reads were aligned to the human reference genome hg19 (UCSC version) using the Burroughs-Wheeler alignment tool.16 The read qualities were recalibrated using GATK Table Recalibration.17 Picard 1.14 was used to flag duplicate reads (http://broadinstitute.github.io/picard/). The GATK IndelRealigner was used to realign the reads around insertion and deletion (InDel) sites.
Single-nucleotide variants (SNVs) were generated with the GATK Unified Genotyper and in parallel with the SAMtools pipeline. The small InDels also were called with the GATK Unified Genotyper and SAMtools.18 The called SNVs and InDels were annotated with ANNOVA.19 The called SNVs and InDels were filtered as follows: (i) exclude high-frequency (minor allele frequency >0.01) polymorphisms in the 1000 Genomes Project and ESP6500; (ii) extract heterozygous variants; (iii) exclude variants in our 100 in-house control samples with exomeSeq data; (iv) extract the segregating variants in the three individuals using whole-exome sequencing; and (v) extract the variants located in the four linkage signal regions. The candidate mutation identified by the above filtering procedures was sequenced by Sanger sequencing in all family members to determine segregation. The detailed whole-exome sequencing method and bioinformatics procedures were described in our previous study.12
Real-time quantitative PCR and immunoblot
For the real-time quantitative PCR (qPCR), total RNA was extracted from the Epstein-Barr virus–transformed lymphoblastoid cell lines with the Thermo GeneJET RNA Purification Kit (Thermo scientific) and was reverse transcribed to complementary DNA with the RevertAidTM First Strand cDNA Synthesis Kit (Thermo scientific). The relative standard curve method was used to analyze the expression of the gene of interest with the MaximaTM SYBR Green/ROX qPCR Master Mix Kit (Fermentas). The expression was normalized to β-actin in the same sample and was measured in three independent real-time qPCRs. The primers were designed using the PRIMER 3 program (http://primer3.ut.ee). The real-time primer pairs for the gene of interest were 5′-CCACTGATGAGGACGAGATAGG-3′ and 5′-CATTGCCTGGTACTTGGTTC-3′; the primer pairs for β-actin were 5′-GGCATGGGTCAGAAGGATT-3′ and 5′-TGGTGCCAGATTTTCTCCA-3′. The reactions were performed using the Bio-Rad CFX96 Touch Deep Well Real-Time PCR Detection System.
For immunoblotting, lymphoblastoid cells from patients and normal controls were washed twice in phosphate-buffered saline and lysed in 2× sodium dodecyl sulfate lysis buffer at room temperature. Equal protein loading was ensured by prior quantitation using the bicinchoninic acid protein assay (Pierce). The protein aliquots were separated by gel electrophoresis in 12% polyacrylamide mini-gels and were transferred onto a polyvinylidene difluoride membrane (Roche Applied Science). Western blot analysis of the protein levels was performed using an antibody produced in rabbits (Sigma). Antibodies against β-actin also were purchased from Sigma.
Mutation screening
The candidate gene identified from whole-genome linkage and whole-exome sequencing was screened by Sanger sequencing in an additional 186 family probands or sporadic cases with high myopia. All exons flanking the splicing sites and the gene untranslated regions were amplified by PCR. The PCR primers were designed with the online Primer3 program (http://frodo.wi.mit.edu/). Sanger sequencing was performed using the ABI 3100/3130 DNA analyzer. All variants identified were confirmed by repetitive, independent PCR amplifications and DNA bidirectional sequencing.
Results
Clinical characterizations
The refractive errors for the five affected subjects from family HM-SR3 ( Figure 1a ) ranged from −6.25 to −10.00 diopter spheres for the left eye and from −6.00 to −20.00 diopter spheres for the right eye. The axial length of the eye globe ranged from 26.14 to 27.45 mm in the left eye and 26.03 to 31.01 mm in the right eye ( Table 1 ). All of the affected individuals had a history of myopia onset before the age of 10 ( Table 1 ) and had no known ocular disease or insult that could predispose them to myopia, such as retinopathy of prematurity or early-age media opacification, and no known genetic disease associated with myopia, such as Stickler syndrome or Marfan syndrome. In addition to high myopia, patient M21370 also presented with late-onset retinal detachment in the left eye. Patient M21355 also presented with slightly late-onset cataracts in both eyes. The additional 186 cases were all early onset (before 10 years old), with refractive errors ranging from −6.0 to −29.0 diopter spheres for both eyes.
Linkage analysis identified candidate linkage regions
To narrow the chromosome intervals and increase the possibility of identifying the causative mutation, whole-genome linkage analysis was performed in HM-SR3. Multipoint parametric linkage analysis identified four candidate regions on chromosomes 1, 5, 6, and 21, with a maximum multipoint parametric log of odds (LOD) score of 2.405 ( Figure 1b and Supplementary Figure S1 online). No other linkage peaks with an LOD score more than 0 was identified. The total size of the four linkage regions was ~69 cM (60 Mb) and included more than 500 genes. Thus, we subsequently used whole-exome sequencing to identify the causative mutation.
Whole-exome sequencing identified a segregating mutation within P4HA2
Whole-exome sequencing was performed on three individuals, including two affected (M21369 and M21458) and one unaffected (M21375) individual ( Figure 1a ) from family HM-SR3. In total, we generated an average of 8.6, 9.73, and 9.91 Gbp of sequence, with more than 50× coverage for each individual as paired-end, 101-bp reads, and more than 98% of the targeted bases were covered sufficiently to pass our thresholds for calling SNVs and InDels (Supplementary Table S2 online).
We identified 22,184 (9,432 nonsynonymous SNVs, splicing SNVs, and InDels), 22,432 (9,474 nonsynonymous SNVs, splicing SNVs, and InDels), and 22,337 (9,467 nonsynonymous SNVs, splicing SNVs, and InDels) coding variants in M21369, M21375, and M21458, respectively. Following the filtering procedures described in the Materials and Methods, only one variant, c.871G>A (p.E291K), within P4HA2 was left for further analysis ( Table 2 , Figure 1c ).We used Sanger sequencing to validate this variant ( Figure 2a ) and analyze the cosegregating status of the variant in all family members. This variant was cosegregated with the phenotype in all family members ( Figure 1a ).

P4HA2 mutations identified in myopia patients. (a) Sequence analyses for the five mutations identified in families with high myopia and sporadic cases. (b) The three missense mutations at position 140 (Q140R), 150 (I150V), and 291 (E291K) of P4HA2 are highly conserved throughout evolution. (c) The pedigree plot and the genotypes for the family with the Q140R mutation. (d) Multipoint parametric linkage analysis combining the two families with P4HA2-segregating mutations demonstrated a significant linkage region in chromosome 5 (LOD = 2.97). (e) Mutation locations in P4HA2 encoded protein identified in high myopia cases. Prolyl 4-hydroxylase, α-polypeptide II contains two important functional domains: a peptide substrate-binding domain and an oxoglutarate/iron-dependent dioxygenase domain. Two tetratricopeptide-like helical regions also are indicated by the underlying green bar. H430, D432, H501, and K511 indicate the critical catalyzing sites.
Variant c.871G>A (p.E291K) is located in the linkage region in chromosome 5, with a maximum multipoint, parametric LOD score of 2.405. To confirm that the segregating status of the mutation was consistent with the haplotype segregating status, we selected a lower-density marker in this region and performed the haplotype analysis using the genotyped single-nucleotide polymorphisms around this mutation. The haplotype analysis demonstrated that the phenotype-segregating variant segregated with the haplotype exactly (Supplementary Figure S2 online). This mutation could not be identified in the 526 population-matched controls using Sanger sequencing or in another cohort of 100 population-matched controls with exome sequencing data.
Segregating mutation is deleterious and produced unstable messenger RNA
The segregating variant c.871G>A (p.E291K) is highly conserved throughout evolution ( Figure 2b , Table 3 ) and is not reported in dbSNP138 and any other larger cohort sequencing data, such as the 1000 Genomes Project or the National Heart, Lung, and Blood Institute ESP exome sequencing projects. To determine the functional consequence of this variant, we first performed functional prediction with SIFT, PolyPhen-2, LRT, and MutationTaster. Functional prediction revealed that this variant is deleterious or possibly damaging ( Table 3 ). To find out whether the missense variant influences the messenger RNA (mRNA) expression level of P4HA2, real-time qPCR was performed on lymphocyte cells from three patients in family HM-SR3. The results revealed that the mRNA expression level was decreased significantly compared with normal controls ( Figure 1d ). With the hypothesis that the missense mutation resulted in unstable mRNA, we used Emetine (dihydrochloride, CAS 316-42-7; Calbiochem), an RNA-degrading inhibitor, to treat the cell line and then performed real-time qPCR. The results demonstrated that there were no differences between the cases and controls ( Figure 1d ). This result indicates that the missense variants may produce unstable, mutated mRNA. We further performed an immunoblot to test the protein level of p4ha2 in familial cases and normal controls. As expected, the protein level in the cases was remarkably decreased compared with the normal controls ( Figure 1e ).
Mutation screening identified multiple new mutations in family or sporadic cases
To identify more genetic evidence to support our findings, we subsequently sequenced the coding exons, flanking splicing sites, and untranslated regions of P4HA2 in an additional 186 subjects with high myopia. Finally, an additional four variants of P4HA2 in five subjects were identified ( Table 3 , Figure 2a , Supplementary Table S1 online). Variant c.419A>G (p.Gln140Arg) was recurrently identified in a family proband (M20438) and a sporadic case (M21457). Variants c.448A>G (p.Ile150Val), c.1327A>G (p.Lys443*), and c.1349_1350delGT (p.Arg451Glyfs*8) each were identified in one sporadic case (M21455, M21812, and M21436, respectively). All of these variants were absent in the 626 controls from the Chinese population with no high myopia or related phenotypes. Functional prediction and conservation analyses revealed that both missense variants (p.Gln140Arg and p.Ile150Val) are deleterious or possibly damaging and are highly conserved throughout evolution ( Table 3 , Figure 2b ).
The missense variant c.419A>G (p.Gln140Arg) was identified in a family proband. This family included six members, and two of them are affected ( Figure 2c ). Sanger sequencing revealed that this variant is segregating with the phenotype in the family ( Figure 2c ). We then performed whole-genome genotyping of this family using the same BeadChip as with family HM-SR3 and performed linkage analysis combining both families. The linkage analysis showed that only the linkage signal, in which P4HA2 is located, demonstrated marginal significant linkage (LOD = 2.97; Figure 2d ).
Discussion
In this study we identified a novel causative gene, P4HA2, for nonsyndromic high myopia. P4HA2 encodes prolyl 4-hydroxylase, α-polypeptide II, which functions as an α2β2 tetramer with a protein disulfide isomerase encoded by P4HB. It contains two important functional domains: a peptide substrate-binding domain and an oxoglutarate/iron-dependent dioxygenase domain20,21 ( Figure 2e ). Two tetratricopeptide-like helical regions located in the substrate-binding domain and the nearby region also were identified ( Figure 2e ). The segregating mutation was located in the second tetratricopeptide-like helical region ( Figure 2e ) and caused unstable mRNA and, consequently, a reduced protein level, as observed by our qPCR and immunoblot experiment. The stopgain and frameshift mutations are both located in the oxoglutarate/iron-dependent dioxygenase domain ( Figure 2e ). The produced truncated mutation deleted the catalytically critical residues, which may result in a loss of function of the mutated allele. The two other missense mutations were located near the substrate-binding domain ( Figure 2e ). These two missense mutations may influence the binding between the enzyme and substrates.
Prolyl 4-hydroxylase, α-polypeptide II, as one of the main isoenzymes, catalyzes the 4-prolyl hydroxylation of collagens. It has been demonstrated that prolyl hydroxylation is essential to the proper three-dimensional folding of newly synthesized procollagen chains for collagens. The nonhydroxylated collagen polypeptide chains cannot form functional molecules in vivo.21 Interestingly, collagens are a primary component of the extracellular matrix of the sclera, and changes in the composition of the extracellular matrix of the sclera have been shown to alter the axial length of the eye.22 Furthermore, reduced collagen accumulation in the sclera is observed in highly myopic eyes.23 Importantly, another collagen prolyl hydroxylase–encoded gene, named LEPREL1, has been reported to cause autosomal-recessive high myopia with early-onset cataracts.13,24 It is also worth noting that several clinical syndromes, such as Stickler syndrome and Marfan syndrome, in which myopia is a prominent characteristic, are caused by defects in collagen. Considering the converged evidence of collagen in high myopia pathogenesis, we hypothesize that mutations of P4HA2 may result in unstable collagens in the sclera, which loses the function to maintain the eye morphology. The disrupted sclera causes the longer axial length of the eye and the high myopia phenotypes.
The limitation of our study is that the participants were limited to the Chinese population. Further research should be performed in other populations to validate whether these results are applicable to patients outside of China. As we stated in the Introduction, known genes causing high myopia explained only a small subset of the patients individually. Similarly, P4HA2 affects only a small subgroup of high myopias, even in the Chinese population. This indicates the high genetic heterogeneity of high myopia, and there may be multiple pathogenetic mechanisms besides the collagen problem.
In summary, we identified mutations in P4HA2 that are associated with high myopia using a combination of whole-genome linkage analysis and whole-exome sequencing and subsequent mutation screening. Our data indicate that the disruption of posttranslational modifications of collagen is an important pathogenetic mechanism for high myopia. Our findings also have implications for future clinical and molecular diagnostics of high myopia. Further research should be conducted to determine the potential pathogenesis driven by mutations in P4HA2, which will be beneficial for developing potential treatment strategies.
Disclosure
The authors declare no conflict of interest.
References
Williams KM, Hammond CJ. Prevalence of myopia and association with education in Europe. Lancet 2014;383:S109.
You QS, Wu LJ, Duan JL, et al. Prevalence of myopia in school children in greater Beijing: the Beijing Childhood Eye Study. Acta Ophthalmol 2014;92:e398–e406.
Morgan IG, Ohno-Matsui K, Saw SM. Myopia. Lancet 2012;379:1739–1748.
Bourne RR, Stevens GA, White RA et al. Causes of vision loss worldwide, 1990–2010: a systematic analysis. Lancet Glob Health 2013;1:e339–349.
Sorsby A, Leary GA, Fraser GR. Family studies on ocular refraction and its components. J Med Genet 1966;3:269–273.
Hysi PG, Wojciechowski R, Rahi JS, Hammond CJ. Genome-wide association studies of refractive error and myopia, lessons learned, and implications for the future. Invest Ophthalmol Vis Sci 2014;55:3344–3351.
Shi Y, Gong B, Chen L, et al. A genome-wide meta-analysis identifies two novel loci associated with high myopia in the Han Chinese population. Hum Mol Genet 2013;22:2325–2333.
Khor CC, Miyake M, Chen LJ, et al.; Nagahama Study Group. Genome-wide association study identifies ZFHX1B as a susceptibility locus for severe myopia. Hum Mol Genet 2013;22:5288–5294.
Shi Y, Li Y, Zhang D, et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet 2011;7:e1002084.
Zhao F, Wu J, Xue A, et al. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum Genet 2013;132:913–921.
Tran-Viet KN, Powell C, Barathi VA, et al. Mutations in SCO2 are associated with autosomal-dominant high-grade myopia. Am J Hum Genet 2013;92:820–826.
Guo H, Jin X, Zhu T, et al. SLC39A5 mutations interfering with the BMP/TGF-ß pathway in non-syndromic high myopia. J Med Genet 2014;51:518–525.
Mordechai S, Gradstein L, Pasanen A, et al. High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am J Hum Genet 2011;89:438–445.
Aldahmesh MA, Khan AO, Alkuraya H, et al. Mutations in LRPAP1 are associated with severe myopia in humans. Am J Hum Genet 2013;93:313–320.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303.
Li H, Handsaker B, Wysoker A, et al.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
Hieta R, Kukkola L, Permi P, et al. The peptide-substrate-binding domain of human collagen prolyl 4-hydroxylases. Backbone assignments, secondary structure, and binding of proline-rich peptides. J Biol Chem 2003;278:34966–34974.
Myllyharju J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med 2008;40:402–417.
Rada JA, Shelton S, Norton TT. The sclera and myopia. Exp Eye Res 2006;82:185–200.
Gentle A, Liu Y, Martin JE, Conti GL, McBrien NA. Collagen gene expression and the altered accumulation of scleral collagen during the development of high myopia. J Biol Chem 2003;278:16587–16594.
Guo H, Tong P, Peng Y, et al. Homozygous loss-of-function mutation of the LEPREL1 gene causes severe non-syndromic high myopia with early-onset cataract. Clin Genet 2014;86:575–579.
Acknowledgements
The authors greatly thank all the patients and their family members who participated in this study. The research was supported by the National Basic Research Program of China (2012CB517902), the National Natural Science Foundation of China (81330027, 81400436, 81161120544, and 31400919) and the Natural Science Foundation of Hunan Province (2015JJ2160).
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
(DOC 356 kb)
Rights and permissions
About this article

Cite this article
Guo, H., Tong, P., Liu, Y. et al. Mutations of P4HA2 encoding prolyl 4-hydroxylase 2 are associated with nonsyndromic high myopia. Genet Med 17, 300–306 (2015). https://doi.org/10.1038/gim.2015.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.28
Keywords
This article is cited by
-
Novel mutations of the X-linked genes associated with early-onset high myopia in five Chinese families
BMC Medical Genomics (2023)
-
Mutational investigation of 17 causative genes in a cohort of 113 families with nonsyndromic early-onset high myopia in northwestern China
Molecular Genetics and Genomics (2023)
-
Fe(2)OG: an integrated HMM profile-based web server to predict and analyze putative non-haem iron(II)- and 2-oxoglutarate-dependent dioxygenase function in protein sequences
BMC Research Notes (2021)
-
Association of 5p15.2 and 15q14 with high myopia in Tujia and Miao Chinese populations
BMC Ophthalmology (2020)
-
Exome genotyping and linkage analysis identifies two novel linked regions and replicates two others for myopia in Ashkenazi Jewish families
BMC Medical Genetics (2019)
