
Abstract
Background/Aims
To determine the genetic basis of myotonia congenita (MC) and strabismus in a large Caucasian family.
Methods
Seven patients making up four generations of a family with MC and strabismus were recruited. All patients had at least one standard ophthalmic examination, including best-corrected visual acuity, refraction, and ocular motility measurements. CLCN1 and SCN4A genes were sequenced and analysed for mutations.
Results
Five out of the seven family members were diagnosed with MC by clinical history and electromyography. Ophthalmic history and exam revealed eyelid myotonia and strabismus. All patients with MC were diagnosed with strabismus between the ages of 3 and 6 and required surgical restoration of ocular alignment. Sequencing results revealed a c. 1333G>A; p. Val445Met mutation in the SCN4A gene.
Conclusion
There are few reports describing eyelid myotonia and strabismus in patients diagnosed with MC. We found significant ocular involvement in a family with a mutation in SCN4A. Future studies may confirm that MC with significant ocular involvement can be used to direct genetic analysis.
Similar content being viewed by others

Introduction
Non-dystrophic myotonic (NDM) syndromes are a heterogeneous group of skeletal muscle disorders that can be caused by mutations in genes encoding chloride (CLCN1) or sodium (SCN4A) channels. CLCN1 and SCN4A encode ion channels in the t-tubules of muscle cells and are important for skeletal muscle fibre contraction.
Mutations in SCN4A can cause a range of sodium channel myotonias, including paramyotonia congenita, hyperkalemic periodic paralysis, and potassium-aggravated myotonias. Mutations in CLCN1 can cause myotonia congenita (MC), a rare disorder with an estimated worldwide prevalence of 1 in 100 000. However, the prevalence varies between geographical regions, with an estimated prevalence in Scandinavia of 1 in 10 000.1 The two major types of MC are Becker disease and Thomsen disease. Thomsen disease is autosomal dominant with early onset and mild myotonia.2 Becker disease is autosomal recessive with late onset and severe myotonia.3
Earlier, only 40–70% of individuals diagnosed clinically with MC were found to harbour the CLCN1 mutation. Recently, a study was conducted on patients with either Thomsen or Becker disease, where patients were screened for mutations in SCN4A in addition to CLCN1. Of all the patients without a mutation in CLCN1, ∼20% had a mutation in SCN4A.4 It has also become apparent that mutations in the two different genes can lead to clinically indistinguishable myotonias, while identical mutations within either gene can also result in a spectrum of clinically heterogeneous phenotypes.5, 6
In previous studies, ocular manifestations of MC have been described, including eyelid myotonia7, 8, 9 and saccade alterations.10, 11 There have been few reports, however, of the disease’s association with strabismus. Wakeman et al11 reported two cases, a father and a son, with Thomsen MC that were evaluated for ocular motility abnormalities. Magnetic resonance imaging revealed ocular muscle hypertrophy and genetic analysis revealed a mutation in SCN4A. Another study described four women from a family with Thomsen disease and associated strabismus, but the disease-causing mutation was not reported.10 In this study, we discuss the largest reported family with both strabismus and MC with a mutation in SCN4A gene.
Materials and methods
Participants and clinical examinations
This study was approved by the Institutional Review Board of the University of California San Diego (UCSD), USA. The subjects were recruited at the Shiley Eye Center and the Abraham Ratner Children’s Eye Center at UCSD. All subjects signed informed consent before participation in this study. Five out of the seven members in the family were diagnosed with MC by clinical history and electromyography. All participants had at least one standard complete ophthalmic examination. Examinations included best-corrected visual acuity measurements, refraction, and ocular motility measurements.
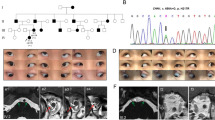
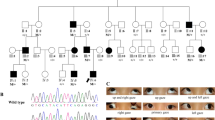
The family being studied consisted of seven Caucasian individuals, including five affected and two unaffected with MC (Figure 1). The proband (IV-1) and her mother (III-3) were seen for their ophthalmic treatments at the UCSD Abraham Ratner Children’s Eye Center. However, the other family members were seen for treatment of their strabismus by different ophthalmologists. Therefore, clinical records from their original diagnosis were not available to us. A thorough history was taken and a current ophthalmic exam was performed.

Pedigree of a family affected by MC. The arrow points to the proband. Results from the genetic analysis of SCN4A are depicted beneath each investigated individual. Only affected individuals (I-2, II-1, III-2, III-3, and IV-1) harbour the V445M mutation.
DNA sequencing
Genomic DNA samples from all seven family members were extracted from peripheral blood leucocytes with the Qiagen kit (Qiagen Inc., Chatsworth, CA, USA) according to the manufacturer’s instructions. Direct nucleotide sequence analysis was completed for both CLCN1 and SCN4A. The 23 exons of CLCN1 and 24 exons of SCN4A were amplified by PCR and sequenced on Genetic Analyzer 3130 (Applied Biosystems, Carlsbad, CA, USA). The primers used to amplify the exons in CLCN1 and SCN4A are presented in Supplementary Table S1.
Results
Clinical examinations
The family consists of seven members, five of which were diagnosed with MC (Figure 1). Clinical history and exams of the five affected family members are presented in Tables 1 and 2. All affected individuals were diagnosed with esotropia between the ages of 3 and 6, and had at least one surgery in both eyes for strabismus. All affected family members reported myotonic symptoms, including muscle stiffness, upon standing and after exercising and improvement of stiffness with repeated muscle contractions. Several individuals described experiencing eyelid myotonia, delayed opening of the eyes after forceful closure. Of note, III-3 also had increased myotonic symptoms during pregnancy and a history of malignant hyperthermia, an associated risk of MC.12
The proband (IV-1) was presented at 5 months of age with esotropia and eyelid myotonia. Other than ocular involvement, she was only mildly affected by myotonia. Despite patching and full cycloplegic spectacles, she developed amblyopia of the left eye by age 3. She subsequently underwent bilateral medial rectus recession and had a positive outcome.
Genetic results
The family history is consistent with an autosomal dominant inheritance pattern (Figure 1). We screened all exons of CLCN1 and SCN4A. All five affected individuals were found to have a G>A transition (c. 1333G>A) in exon 9 of SCN4A gene, resulting in a V445M substitution. This variation was absent in the two unaffected family members (Figure 2). The change (c. 1333G>A) was also absent in 1000 normal control chromosomes. No novel or previously reported mutations were found in CLCN1 gene in all the affected family members.

The SCN4A sequencing results. Sequencing data show that a G–A transition in exon 9 (arrow) causes the V445M mutation in affected individuals (top). This alteration was absent in the unaffected individuals (bottom).
Discussion
We report here clinical features and genetic analysis of the largest family with MC and strabismus. We initially screened for mutations in CLCN1 based on reports of increased frequency of mutations within this gene in patients with MC.1, 4 However, as no mutations were found, we screened for mutations in SCN4A. SCN4A is located on 17q23 and includes 35 kb with 24 exons.13 All affected family members were found to have a G>A transition within exon 9 of SCN4A, resulting in the V445M mutation. This mutation has been associated with debilitating, painful myotonia, and muscle hypertrophy.14, 15, 16 However, the family we studied did not have these findings, suggesting that other modifying factors may be involved in determining the above-mentioned phenotype.
Results from multiple studies demonstrate that patients with SCN4A mutations are more likely to have ocular symptoms, either eyelid myotonia or strabismus.8, 9, 11 One study showed that 96.7% of patients with sodium channel mutations had eyelid myotonia compared with 46.9% of patients with chloride channel mutations. Furthermore, mutations in SCN4A cause more severe eyelid myotonia than mutations in CLCN1.8 An increased number of sodium channels in facial muscles compared with other skeletal muscles may explain the findings associated with SCN4A mutations.8, 17, 18 Consistent with our analysis, the only other genetic study performed on patients with strabismus in addition to myotonia also found the V445M mutation.19 There may be an association between this mutation and extraocular muscles. It has been speculated that esotropia may be caused by a slightly nasally displaced superior and inferior rectus, as well as an inferiorly displaced lateral rectus.11 However, imaging of a larger cohort with more detailed scans are necessary to confirm an association between the mutation and such anatomic anomalies.
In conclusion, even though this is a rare condition, ophthalmologists should be aware of its ocular manifestations. In our study, eyelid myotonia and strabismus in the proband led to the suspicion of MC. As these two ocular findings can present as early as 5–6 months of age, ophthalmologists may be the first to evaluate patients with NDM. Ophthalmologists should be aware of the reported risk of malignant hyperthermia in patients with MC12 and alert the anaesthesiologist before the use of general anaesthesia. Finally, our results may support the sequencing of SCN4A before CLCN1 in myotonic patients with significant ocular involvement.

References
Sun C, Tranebjaerg L, Torbergsen T, Holmgren G, van Ghuelue M . Spectrum of CLCN1 mutations in patients with myotonia congenita in northern Scandinavia. Eur J Hum Genet 2001; 9: 903–909.
Thomsen J . Tonische Krampfe in willkurlich beweglichen Muskelm in Folge von ererbter psychischer disposition (ataxia muscularis?). Arch Psychiatr Nervenkr 1876; 6: 702–718.
Becker PE . Myotonia Congenital and Syndromes Associated with Myotonia. Thieme: Stuttgart, 1977.
Trip J, Drost G, Verbove DJ, van der Kooi AJ, Kuks JB, Notermans NC et al. In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotonia. Eur J Hum Genet 2008; 16: 921–929.
Colding-Jorgenson E . Phenotypic variability in myotonia congenital. Muscle Nerve 2005; 32: 19–34.
Lee SC, Kim HS, Park YE, Choi YC, Park KH, Kim DS . Clinical diversity of SCN4A-mutation-associated skeletal muscle sodium channelopathy. J Clin Neurol 2009; 5: 186–191.
Davidson S . The eye in dystrophia myotonica with a report on electromyography of the extra-ocular muscles. Brit J Ophthal 1961; 45: 183–196.
Trip J, Drost G, Ginjaar I, Nieman F, van der Kooi AJ, de Visser M et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry 2009; 80: 647–652.
Stunnenberg BC, Ginjaar HB, Trip J, Faber CG, van Engelen BG, Drost G . Isolated eyelid closure myotonia in two families with sodium channel myotonia. Neurogenetics 2010; 11: 257–260.
Spire M, Tilikete C, Habault C, Vial JC, Kodjikian L, Grange JD . Thomsen myotonia congenita and strabismus: recording saccades in myotonic diseases. J Fr Ophtalmol 2006; 29 (3): 271–280.
Wakeman B, Babu D, Tarleton J, MacDonald I . Extraocular muscle hypertrophy in myotonia congenita. J AAPOS 2008; 12: 294–296.
Rossenbaum H, Miller J . Malignant hyperthermia and myotonic disorders. Anesthesiol Clin North America 2002; 20 (3): 623–664.
George AL, Ledbetter DH, Kalen RG, Barchi RL . Assignment of a human skeletal muscle sodium channel alpha-subunit gene (SCN4A) to 17q23.1-25.3. Genomics 1991; 9 (3): 555–556.
Rosenfeld J, Sloan-Brown K, George AL . A novel muscle sodium channel mutation causes painful congenital myotonia. Ann Neurol 1997; 42: 811–814.
Dupre N, Chrestian N, Bouchard JP, Rossignol E, Brunet D, Sternberg D et al. Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscul Disord 2009; 19: 330–334.
Wang DW, VanDeCarr D, Ruben P, George AL, Bennett PB . Functional consequences of a domain 1/S6 segment sodium channel mutation associated with painful congenital myotonia. FEBS Letters 1999; 448: 231–234.
Caldwell JH . Clustering of sodium channels at the neuromuscular junction. Microsc Res Tech 2000; 49: 84–89.
Happak W, Liu J, Burggasser G, Flowers A, Gruber H, Freilinger G . Human facial muscles: dimensions, motor endplate distributions, and presence of muscle fibers with multiple motor endplates. Anat Rec 1997; 249: 276–284.
Wakeman B, MacDonald IM, Ginjaar I, Tarleton J, Babu D . Extraocular muscle hypertrophy in myotonia congenital: mutation identified in the SCN4A gene (V445M). J AAPOS 2009; 13 (5): 526–527.
Acknowledgements
We acknowledge Maureen Crocker, Bin Lin, Karen Ysensky and Dr Salma Khayali for their help in this study. KZ is supported by grants from the NEI/NIH (EY014428, EY018660, and EY019270), VA Merit Award, Research to Prevent Blindness, and BWF Clinical Scientist Award in Translational Research. SG is supported by the UCSD Clinical and Translational Research Institute 1TL1RR031979-01.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Eye website
Supplementary information
Rights and permissions
About this article
Cite this article
Du, H., Grob, S., Zhao, L. et al. Myotonia congenita with strabismus in a large family with a mutation in the SCN4A gene. Eye 26, 1039–1043 (2012). https://doi.org/10.1038/eye.2012.80
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2012.80
