
Abstract
Cerebellar dysplasia with cysts and abnormal shape of the fourth ventricle, in the absence of significant supratentorial anomalies and of muscular involvement, defines recessively inherited Poretti–Boltshauser syndrome (PBS). Clinical features comprise non-progressive cerebellar ataxia, intellectual disability of variable degree, language impairment, ocular motor apraxia and frequent occurrence of myopia or retinopathy. Recently, loss-of-function variants in the LAMA1 gene were identified in six probands with PBS. Here we report the detailed clinical, neuroimaging and genetic characterization of 18 PBS patients from 15 unrelated families. Biallelic LAMA1 variants were identified in 14 families (93%). The only non-mutated proband presented atypical clinical and neuroimaging features, challenging the diagnosis of PBS. Sixteen distinct variants were identified, which were all novel. In particular, the frameshift variant c.[2935delA] recurred in six unrelated families on a shared haplotype, suggesting a founder effect. No LAMA1 variants could be detected in 27 probands with different cerebellar dysplasias or non-progressive cerebellar ataxia, confirming the strong correlate between LAMA1 variants and PBS.
Similar content being viewed by others


Introduction
Cerebellar malformations comprise a wide range of developmental defects of the cerebellum that are often associated with morphological anomalies of the brainstem, present typically within the first month of life, and have a non-progressive course. For some cerebellar malformations such as Joubert syndrome (JS), Dandy–Walker malformation and rhombencephalosynapsis, specific diagnostic criteria have been established based on neuroimaging findings, and the diagnosis relies purely on the unique neuroimaging phenotype.1, 2, 3 Other cerebellar malformations, however, are characterized by less specific cerebellar imaging findings such as hypoplasia, dysplasia and cysts.4, 5, 6 These findings may be isolated or occur as part of more complex diseases, and their diagnostic definition is more challenging. In the last decade, progress and increasing experience with neuroimaging techniques and genetic analysis have greatly improved the diagnostic yield of cerebellar malformations, thanks to the better neuroimaging-based delineation and the identification of several causative genes. In a large proportion of patients, however, the underlying cerebellar malformations remains poorly defined.
Recently, we delineated a novel autosomal recessive cerebellar malformations that is characterized on neuroimaging by cerebellar dysplasia with cysts (CDC), with an enlarged, elongated and square-like shaped fourth ventricle.7 Additional common findings include hypoplasia of the cerebellar vermis and mild brainstem abnormalities such as elongated midbrain or short pons, whereas malformations of cortical development are consistently absent. The clinical phenotype is also rather homogeneous: all the seven children originally described presented non-progressive cerebellar ataxia, intellectual disability of variable degree and language impairment, without any muscular involvement. Ocular motor apraxia and myopia were also common.7 This novel disease was subsequently termed Poretti–Boltshauser syndrome (PBS, MIM #615960).8 The lack of significant supratentorial anomalies and of muscular involvement clearly distinguish PBS from alpha-dystroglycanopathies and GPR56-related bilateral frontoparietal polymicrogyria that are both characterized by frequent CDC.5
In 2014, variants in the LAMA1 gene (MIM*150320) were identified in an independent cohort of five probands with PBS, and in a sixth family presenting mild cerebellar dysplasia without cysts.8 Motor delay, abnormal ocular movements and myopia were detected in all patients, whereas language delay and intellectual disability were inconsistent findings. In addition, all patients presented some degree of retinal involvement including full-blown retinal dystrophy in some of them. From a neuroimaging point of view, besides the typical findings, some of the patients had splayed superior cerebellar peduncles (SCP) and mild, patchy T2/FLAIR-hyperintense signal in the periventricular white matter, without obvious clinical relevance. Identified variants included nonsense, frameshift, splice site variants and multiexon deletions, all predicted to result in a complete loss of function of the protein.8
Here we report the results of LAMA1 variant analysis in a large cohort of 15 probands with PBS. In addition, to assess the specificity of LAMA1-associated clinical and neuroimaging phenotype, we searched for LAMA1 variants in 27 probands with distinct isolated cerebellar dysplasias or other forms of non-progressive cerebellar ataxia.
Patients and Methods
This study was approved by the ethics committee of the IRCCS Casa Sollievo della Sofferenza. Written informed consent was obtained from all families.
Patient cohorts
The 15 probands with PBS were collected by the senior authors (EBo and EMV): (1) from their personal cohorts of patients; (2) from patients who were referred for second opinion; (3) from requests to evaluate clinical and neuroimaging data of patients with CDC; and (4) from patients referred for molecular genetic testing after the diagnosis of PBS. Five probands were included in the original publication.7 The inclusion criteria were as follows: (1) a clinical phenotype compatible with PBS including non-progressive cerebellar ataxia, ocular motor apraxia, intellectual disability, myopia and/or retinal dystrophy and (2) cerebellar dysplasia with or without cysts.7 Exclusion criteria were as follows: (1) genetically confirmed or suspected α-dystroglycanopathies (muscular involvement with elevated CK and/or suggestive malformations of cortical development such as cobblestone brain); (2) genetically confirmed or suspected GPR56-related polymicrogyria; (3) genetically confirmed or suspected tubulinopathies (dysmorphic basal ganglia); and (4) unilateral cerebellar dysplasia suggestive of an acquired, disruptive etiology.
In addition, as part of a next-generation-sequencing-based panel for cerebellar malformations or non-progressive cerebellar ataxia, LAMA1 was also sequenced in a second cohort of 27 probands with either cerebellar dysplasias not matching the diagnostic criteria for PBS (n=5) or non-progressive cerebellar ataxia with cerebellar hypoplasia and enlarged interfoliar spaces (n=22). Probands with JS were not included in this screening, as Aldinger and collaborators had previously excluded LAMA1 variants in a large cohort of JS subjects.8
Clinical and qualitative neuroimaging analysis
Demographic data and detailed information about neurological and ophthalmological features as well as neurodevelopmental outcome were collected by (1) review of the clinical histories and (2) clinical-neurological follow-up examinations. Whenever possible, cognitive and language functions have been assessed with standardized neuropsychological tests as appropriate for the age of the patients, to provide an intelligence quotient (IQ) or developmental quotient. Otherwise, the developmental stage was estimated from the patient’s history, clinical observations and kindergarten or school reports.
All available neuroimaging data sets were retrospectively analyzed by two pediatric neurologists with experience in neuroimaging of the pediatric cerebellum (AP and EBo). Detailed information about neuroimaging analysis is described in Supplementary Methods.
LAMA1 sequencing analysis
In all probands, LAMA1 was sequenced as part of a panel of 44 genes causative of various cerebellar malformations or non-progressive cerebellar ataxia (with the exclusion of JS genes), with TruSeq Custom Amplicon (TSCA) technology on a MiSeq platform (Illumina, San Diego, CA, USA), following the manufacturer’s protocol. Expected coverage was 99% of the targeted genomic regions of interest. Sequences were mapped against the hg19 reference genome by Bowtie2, which achieved an average alignment performance of 98% across all samples. Variants were called by the HaplotypeCaller tool of GATK ver. 4.3 (Cambridge, MA, USA), and were annotated with ANNOVAR. Annotated data were filtered out to exclude intronic variants, synonymous variants not predicted to affect splice sites, as well as variants with reported frequency ≥0.01 in publicly available human variation resources (dbSNP ver. 144, 1000 Genomes, Exome Aggregation Consortium (ExAC), NHLBI Exome Sequencing Project Exome Variant Server (EVS)). Missense variants surviving the previous filters were investigated using dedicated software to assess their pathogenicity (PolyPhen-2, Boston, MA, USA; SIFT, La Jolla, CA, USA; MutationAssessor, New York, NY, USA; MutationTaster, Berlin, Germany and CADD, University of Washington, Seattle, WA, USA, and HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA) and conservation (phyloP, New York, NY, USA; GERP++, Stanford, CA, USA; SiPhy, Cambridge, MA, USA and ClustalW2, Cambridge, UK). Variants potentially affecting function were verified by Sanger sequencing in the whole nuclear family, to assess segregation with the disease. Accession numbers are as follows: human LAMA1 genomic sequence: NG_034251.1; human LAMA1 mRNA sequence: NM_005559.3; human LAMA1 protein sequence: NP_005550.2. Exons were numbered as in Aldinger et al.8 In two probands in whom only a single heterozygous LAMA1 variant could be identified, sequencing data were further inspected to search for potential heterozygous deletions involving one or more exons. To this aim, a custom script tool was used to check and identify any significant difference in coverage for the LAMA1 amplicons between the two probands and other 82 subjects analyzed with the same panel (including healthy controls and probands with distinct cerebellar malformations). All identified variants have been submitted to the LOVD gene variant database. Software websites are listed in the 'Online resources' section of the Supplementary Methods.
Quantitative real-time PCR
Dosage analysis of LAMA1 exons 31–32 and 62–63 was performed in two probands (patients 10 and 14, respectively), their parents and one healthy control by quantitative real-time PCR, using SYBR green I dye chemistry and a 7900HT sequence detection platform (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). Primers’ sequences are available upon request. The telomerase housekeeping gene was amplified as internal control, and all reactions were run in triplicate. The relative expression between housekeeping gene and each target exon was calculated using the ΔΔCT method.
Haplotype analysis
LAMA1 variant c.[2935delA] was identified in six unrelated families. To search for a common haplotype, five microsatellite markers spanning about 3 Mb around the LAMA1 locus were genotyped in available family members. Fluorescined PCR products were run on an ABI3130XL Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), and raw data were analyzed using the GeneMapper Software Version 4.0 (Applied Biosystems, Thermo Fisher Scientific) to obtain the allele repeat in base pairs. Data analysis was performed with the GeneMarker software (SoftGenetics, State College, PA, USA). Haplotypes were constructed including these five markers as well as 16 single-nucleotide polymorphisms (SNPs) obtained from LAMA1 sequencing data, assigning phase based on the minimum number of recombinants.
Results
Identification of LAMA1 variants
NGS analysis in 15 PBS probands identified biallelic LAMA1 variants in 12, and single heterozygous variants in two. In the latter subjects, comparative analysis of LAMA1 amplicons revealed a significantly reduced coverage of amplicons related to exons 31–32 (patient 10) and exons 62–63 (patient 14). This suggested the presence of heterozygous multiexon deletions, which were confirmed in both cases by quantitative real-time analysis on genomic DNA (Supplementary Figure 1). Thus, homozygous or compound heterozygous variants were detected in 14 out of 15 probands with PBS (93%; see Supplementary Table 1 for details of identified variants).
Overall, we identified 16 distinct variants, all of which were novel. These include five small indels predicting premature truncation of the protein, five nonsense variants, two missense variants, two variants affecting canonical splice sites and two multiexon deletions (Figure 1). The two missense changes were found in compound heterozygosity in patient 15: they affected fully conserved residues and were both predicted as deleterious by four distinct in silico predictors (Supplementary Figure 2). Twelve variants were absent from public databases, whereas four were present either in ExAC or EVS with very low frequency (<0.01), but never in the homozygous state. Segregation with the disease was confirmed in all families. Interestingly, the LAMA1 c.[2935delA] variant was found to recur in six unrelated PBS families (homozygous in five and compound heterozygosity with another frameshift variant in one). This variant was found in the ExAC database with a very low frequency of 8.2e−06, whereas it is not reported in EVS. To evaluate whether this could represent a founder variant, we reconstructed haplotypes spanning the LAMA1 locus, including microsatellite markers and informative intragenic SNPs. Haplotype analysis identified a small region of ~55 Kb shared by all families, between flanking SNPs rs74938552:G>A and rs677136:T>G, suggesting an ancestral origin (Supplementary Table 2).

Schematics of LAMA1 gene and LAMA1 protein with identified variants. Schematic structure of (a) LAMA1 gene and (b) LAMA1 protein. In the gene, vertical black bars denote the 63 exons. In the protein, SMART predicted domains are represented. Variants above and below the panels are those identified in this paper and by Aldinger et al, respectively. Splice site variants are not reported at the protein level. Horizontal gray bars denote multiexon deletions (a) or amino-acid deletions (b). The full colour version of this figure is available at European Journal of Human Genetics online.
No LAMA1 variants could be detected in 27 probands with distinct cerebellar dysplasia or non-progressive cerebellar ataxia phenotypes, confirming the strong correlate between LAMA1 variants and PBS.
Patients’ characteristics and clinical findings
Overall, LAMA1 variants were identified in 17 patients from 14 unrelated families. Clinical findings of these mutated patients are summarized in Table 1. Pregnancy, delivery and perinatal history were uneventful. All children presented within the first 6 months of life mostly with developmental delay or with abnormal ocular movements (strabismus, ocular motor apraxia and nystagmus) and poor fixation. At the last follow-up at the mean age of 8 years (range 1.5–16 years), all children older than 2 years (n=16) had cerebellar ataxia. Eye movement disorders included ocular motor apraxia in 13 children (76%), strabismus in 7 (41%) and nystagmus in 5 (29%). All patients but two (88%) had mild or moderate intellectual disability or cognitive delay and delayed speech/language development. Cognitive functions were assessed by standardized cognitive tests in four patients (patient 1-8-15-17), resulting in widely variable full IQ scores (70, 69, 118 and 32, respectively). Other significant neurological findings were present only in patient 10 who had ballistic movements. Eye involvement was present in 10 patients (59%), including myopia in 9 (53%), hypermetropia in 2 (12%) and retinal dystrophy in 4 (24%) as judged by fundoscopy. No further characterization of retinal involvement by electroretinography or optical coherence tomography was available. None of the mutated patients had systemic involvement outside the brain or eyes.
In patient 18, point and small indel variants were excluded, and there were no hints of reduced coverage in LAMA1 by NGS analysis. At the age of 14 years, this patient had non-progressive cerebellar ataxia, ocular motor apraxia and strabismus, mild cognitive and language impairment, and hypermetropia. In addition, she developed kyphosis in the first year of life that remained stable for ~10 years and then complicated with a severe progressive scoliosis. Detailed clinical information of mutated and non-mutated PBS patients are shown in Supplementary Table 1.
Qualitative neuroimaging findings
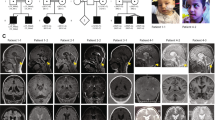
Qualitative neuroimaging analysis of the 17 LAMA1-mutated patients at the mean age at MRI of 3.1 years (range 15 days to 14 years) are summarized in Table 2, and representative images are shown in Figures 2a–f. Cortical-subcortical cerebellar cysts were present in all children and involved both cerebellar hemispheres and the cerebellar vermis in 16 patients (the youngest patient with cerebellar cysts was 3 months old at brain MRI). In the majority of patients, cysts were located within the anterior and superior part of the vermis and the posterior and superior part of the cerebellar hemispheres. Cerebellar dysplasia involved the whole cerebellum in 16 children. In patient 7, however, dysplasia was limited to the inferior parts of the cerebellum, and small cysts were present only in the right cerebellar hemisphere and vermis.

Typical and atypical neuroimaging findings of PBS. (a–d) Typical neuroimaging findings of PBS as seen in 16 patients. (a) Midsagittal T1-, (b, c) axial T2- and (d) coronal T2-weighted magnetic resonance images of patient 10 at the age of 1 year and 5 months show an enlarged, elongated and square-like formed fourth ventricle (asterisk), dysplasia and multiple cortical-subcortical cysts within the cerebellar vermis and both cerebellar hemispheres, and splayed superior cerebellar peduncles (arrows). (e–h) Atypical neuroimaging findings of PBS as seen in two patients. (e) Midsagittal and (f) coronal T2-weighted magnetic resonance images of patient 7 at the age of 8 years and 4 months show mild cerebellar dysplasia involving the inferior parts of the cerebellum, small cysts within the right cerebellar hemisphere (arrow) and vermis (arrowhead), and a normal, triangular shape of the fourth ventricle. (g) Axial and (h) coronal T2-weighted images of patient 18 at the age of 14 years show a global cerebellar dysplasia without cysts, but a slit that is isointense to cerebrospinal fluid and is located within the bilateral cerebellar hemispheres laterally to the middle cerebellar peduncles (arrows). In addition, the superior cerebellar peduncles are only mildly splayed.
Hypoplasia of the cerebellar vermis was found in 10 patients (59%) and affected mostly the inferior part of the vermis. The fourth ventricle was enlarged in 14 children (82%) and had an elongated, square-like form in 13 (77%). On coronal images, the SCP course was splayed in nine children (53%). Morphological abnormalities of the brainstem were seen in six patients (35%) and included an elongated midbrain in six and a short pons in three children. Supratentorial anomalies were seen only in two children (12%) who had mild ventriculomegaly, whereas white matter signal abnormalities or disorders of cortical development were not present in any patient.
Patient 18 showed global cerebellar dysplasia, enlarged, elongated and squared-like formed fourth ventricle, and mild brainstem anomalies; however, she did not have obvious cerebellar cysts, but a slit filled by cerebrospinal fluid within the bilateral cerebellar hemispheres laterally to the middle cerebellar peduncles. The SCP were normally formed (Figures 2g and h).
Discussion
In 2014, we described a novel syndrome characterized by CDC and a homogeneous clinical presentation. Subsequently, Aldinger and collaborators identified variants in the LAMA1 gene in six probands with this syndrome, which they termed PBS.
Here we reported a detailed clinical, neuroimaging and genetic characterization of the largest cohort of PBS patients to date (n=18), detecting LAMA1 biallelic variants in all but one. All 16 identified variants were novel and were scattered throughout the coding region of the gene, without hotspots. In line with the previous report,8 nearly all identified variants were frameshift, nonsense, splice site variants or multiexon deletions, predicted to result in complete loss of function of the protein. Only two missense variants were found in compound heterozygosity in the same subject (patient 15). This was one of two patients in our cohort who had normal cognitive functions at age 9 years (full IQ 118 and 99), despite showing typical neuroimaging findings of PBS. However, the other patient with normal cognitive function (patient 8) carried two variants both predicted to result in truncated protein products, arguing against genotype–phenotype correlations in LAMA1-associated disease and suggesting the existence of other, still unknown phenotypic modifiers. In line with this, Aldinger and collaborators also reported two siblings with normal intelligence (UW160-3 and -4), who were compound heterozygous for a truncating variant and two splice site changes both inherited from the same parent, which effect at the protein level has not been elucidated.8
Six out of 15 families shared the same c.[2935delA] variant (Supplementary Table 1). All these families originated from countries related to the Mediterranean area (Albania, Kosovo, Bosnia, Spain and Turkey) and shared a very small haplotype across the LAMA1 locus, suggesting an ancestral founder effect.
The identification of LAMA1 variants in all but one families confirms the identity of PBS as a well-defined Mendelian disease, and has helped to delineate its clinical and imaging spectrum. The clinical presentation is characterized by non-progressive cerebellar ataxia, intellectual disability and in some children eye movement disorders. This phenotype is rather consistent but non-specific, and hence does not provide useful clues for the diagnosis, especially at onset. In fact, all patients presented within the first 6 months of life with unspecific neurological signs such as hypotonia and delayed motor, and language developmental milestones or with early visual problems. Later on, most patients developed mild-to-moderate intellectual disability. School performance of these children is also hampered by additional issues such as eye movement disorders (eg, ocular motor apraxia), coordination problems (eg, dysmetria) and visual impairments (eg, high myopia and/or retinal dystrophy). However, several children were able to attend regular classes with individual educational support. As previously discussed, in rare patients, cognitive functions may be normal; yet, the degree of intellectual impairment does not seem to correlate with the severity of cerebellar and brainstem malformation.
Ocular involvement in the form of high myopia or retinal abnormalities also occurs frequently in LAMA1-mutated patients. This is in line with findings in Lama1 mouse and zebrafish models that showed defects in the retinal inner limiting membrane, attenuated electroretinography, abnormal lens and retinal vasculature, and progressive cell loss from the inner nuclear and ganglion cell layers. These findings suggest a role for LAMA1 in the basement membranes not only of the mesenchyme adjacent to the developing cerebellum but also of the retina.9, 10, 11, 12 Extra-retinal eye involvement is also present in other ‘laminopathies’: optic atrophy has been reported in children with LAMB1 variants and microcoria, hypoplasia of the iris and ciliary body, whereas lenticonus posterior has been found in LAMB2-mutated patients.13, 14
Although the clinical presentation of PBS can be rather unspecific, brain MRI clearly points to the correct diagnosis by showing the peculiar association of cerebellar dysplasia, cerebellar cysts and abnormally shaped/sized fourth ventricle. Cerebellar dysplasia affects typically the entire cerebellum, although it may be limited only to the superior or inferior parts in rare cases (patient 7 in our series and patients UW163-3 and UW163-4 in Aldinger et al.8). The presence of cerebellar dysplasia in LAMA1-mutated patients is in line with the cerebral phenotype of Lama1 conditional knockout mice.15, 16 In these animal models, study of the postnatal cerebellum revealed misplacing of granule cells that may explain the overall perturbation of the layering of the cerebellum, and an aberrant folia formation that resembled human cerebellar dysplasia.15
Multiple cortical and subcortical cysts are usually found within the superior and anterior vermis, and the superior and posterior parts of both cerebellar hemispheres; however, they may vary in terms of number, size and location. On the basis of the role of laminin α1 in the basement membrane, a similar pathogenesis can be postulated for the cerebellar cysts observed in PBS and in α-dystroglycanopathies: cysts are most likely formed from the subarachnoid spaces that were engulfed by the dysplastic cerebellar folia, particularly in the boundary between the normal and dysplastic cerebellar cortex.17 No cysts were found in the same two siblings from family UW163; however, both patients were young at MRI (5 and 25 months old, respectively), and it is still unknown whether cerebellar cysts in PBS are already present in the neonatal period or develop later, as observed in α-dystroglycanopathies.18, 19 Longitudinal MRI studies of LAMA1-mutated patients starting during the neonatal period are needed to definitely answer this question. All other imaging findings are present and identifiable at birth. To date, the lack of cerebellar cysts within the first week of life does not exclude the diagnosis of PBS in infants with global cerebellar dysplasia, and a brain MRI follow-up after several months should be considered.
In the majority of LAMA1-mutated patients, the fourth ventricle is enlarged and has a peculiar, elongated and square-like form. To the best of our knowledge, this form of the fourth ventricle is unique for PSB and, if present, increases the specificity of the neuroimaging findings. Moreover, an abnormal, splayed and elevated course of the SCP can be observed in about half of patients. The molar tooth sign (MTS), which represents the unique neuroimaging diagnostic criterion of JS, is also characterized by an abnormal course of the SCP.20, 21 However, the appearance of the MTS (elevated, horizontal and thickened SCP) is clearly different, and distinction between PBS and JS is not problematic. Indeed, we are not aware of a single patient with the MTS and cerebellar cysts, and no variants in LAMA1 could be identified in a large cohort of patients with JS.8
Hypoplasia of the inferior cerebellar vermis and mild abnormalities of the brainstem have been found in the majority of children with PBS. Aldinger and collaborators also reported occasional supratentorial abnormalities, including patchy T2-hyperintense signal of the periventricular white matter in three patients and mild ventriculomegaly in one patient.8 We did not find white matter signal abnormalities in our patients, but mild ventriculomegaly in two. All these represent inconsistent and non-specific findings that are not helpful for diagnostic purposes. Generally, the presence of significant morphological abnormalities or signal changes within the supratentorial brain should question the diagnosis of PBS.
Although the phenotype of LAMA1-mutated patients is fairly homogeneous, we observed some unexpected intrafamilial variability: in a large family with four affected siblings (patients 4–7, Supplementary Table 1), one child had different, unusual neuroimaging findings compared with his sibs, including only few small cysts, normal size and shape of the fourth ventricle, and normal morphology of the brainstem. This observation suggests the existence of still unknown modifiers able to modulate the phenotypic expression even in patients carrying the same gene variants, as shown in other cerebellar malformations such as JS.21, 22
In one patient, we diagnosed PBS based on an early low-quality MRI at age 5 months, but failed to identify variants affecting function or exon imbalances in LAMA1. Interestingly, this patient seems to be an outlier both from a clinical and neuroimaging point of view. In fact, she presented a severe kyphoscoliosis since infancy that has not been found in any of the other PSB patients; moreover, a recent, higher-quality MRI at age 14 years did not show typical cerebellar cysts, but bilateral CSF isointense slits within the cerebellar hemispheres laterally to the middle cerebellar peduncles, which could represent the fusion of multiple small cysts (Figures 2g and h). Although we cannot rule out the possibility that this patient carries LAMA1 variants not detectable with the adopted methods (eg, deep intronic variants or promoter variants), the atypical clinical and neuroimaging features argue against this option, and support the observation that the LAMA1-associated phenotype is highly homogeneous and well recognizable. Indeed, other conditions characterized by the presence of cerebellar dysplasia and/or cerebellar cysts (such as α-dystroglycanopathies, GPR56-related polymicrogyria, tubulinopathies, Chudley–McCullough syndrome, Aicardi syndrome or some forms of ponto-cerebellar hypoplasia) can be easily ruled out by the presence of additional, peculiar neuroimaging findings (eg, specific malformations of cortical development, dysmorphic basal ganglia, corpus callosum anomalies and severe pons hypoplasia) and/or clinical features (eg, muscular dystrophy, spasticity, severe epilepsy and so on), making the differential diagnosis of PBS relatively easy.5, 7
In patients with suspicion of PBS, LAMA1 genetic analysis is warranted to confirm the diagnosis and provide genetic counseling. As PBS appears to be a genetically homogeneous condition and the LAMA1 gene is very large and lacks hotspots, testing for the founder variant c.[2935delA] might be considered as a first, rapid and inexpensive diagnostic step, as this variant is likely responsible for at least 40% of PBS cases originated from the Mediterranean area. In the remaining patients, NGS-based sequencing of LAMA1 would be preferable over Sanger sequencing, as it will also allow identifying heterozygous exon deletions.
References
Doherty D, Millen KJ, Barkovich AJ : Midbrain and hindbrain malformations: advances in clinical diagnosis, imaging, and genetics. Lancet Neurol 2013; 12: 381–393.
Bosemani T, Orman G, Boltshauser E, Tekes A, Huisman TA, Poretti A : Congenital abnormalities of the posterior fossa. Radiographics 2015; 35: 200–220.
Jissendi-Tchofo P, Severino M, Nguema-Edzang B, Toure C, Soto Ares G, Barkovich AJ : Update on neuroimaging phenotypes of mid-hindbrain malformations. Neuroradiology 2015; 57: 113–138.
Poretti A, Boltshauser E, Doherty D : Cerebellar hypoplasia: differential diagnosis and diagnostic approach. Am J Med Genet C Semin Med Genet 2014; 166C: 211–226.
Boltshauser E, Scheer I, Huisman TA, Poretti A : Cerebellar cysts in children: a pattern recognition approach. Cerebellum 2015; 14: 308–316.
Poretti A, Boltshauser E : Terminology in morphological anomalies of the cerebellum does matter. Cerebellum Ataxias 2015; 2: 8.
Poretti A, Hausler M, von Moers A et al: Ataxia, intellectual disability, and ocular apraxia with cerebellar cysts: a new disease? Cerebellum 2014; 13: 79–88.
Aldinger KA, Mosca SJ, Tetreault M et al: Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Am J Hum Genet 2014; 95: 227–234.
Edwards MM, Mammadova-Bach E, Alpy F et al: Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J Biol Chem 2010; 285: 7697–7711.
Edwards MM, McLeod DS, Grebe R, Heng C, Lefebvre O, Lutty GA : Lama1 mutations lead to vitreoretinal blood vessel formation, persistence of fetal vasculature, and epiretinal membrane formation in mice. BMC Dev Biol 2011; 11: 60.
Biehlmaier O, Makhankov Y, Neuhauss SC : Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest Ophthalmol Vis Sci 2007; 48: 2887–2894.
Semina EV, Bosenko DV, Zinkevich NC et al: Mutations in laminin alpha 1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev Biol 2006; 299: 63–77.
Radmanesh F, Caglayan AO, Silhavy JL et al: Mutations in LAMB1 cause cobblestone brain malformation without muscular or ocular abnormalities. Am J Hum Genet 2013; 92: 468–474.
Wuhl E, Kogan J, Zurowska A et al: Neurodevelopmental deficits in Pierson (microcoria-congenital nephrosis) syndrome. Am J Med Genet A 2007; 143: 311–319.
Heng C, Lefebvre O, Klein A et al: Functional role of laminin alpha1 chain during cerebellum development. Cell Adh Migr 2011; 5: 480–489.
Ichikawa-Tomikawa N, Ogawa J, Douet V et al: Laminin alpha1 is essential for mouse cerebellar development. Matrix Biol 2012; 31: 17–28.
Aida N, Yagishita A, Takada K, Katsumata Y : Cerebellar MR in Fukuyama congenital muscular dystrophy: polymicrogyria with cystic lesions. AJNR Am J Neuroradiol 1994; 15: 1755–1759.
Barkovich AJ : Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol 1998; 19: 1389–1396.
Longman C, Mercuri E, Cowan F et al: Antenatal and postnatal brain magnetic resonance imaging in muscle-eye-brain disease. Arch Neurol 2004; 61: 1301–1306.
Maria BL, Hoang KB, Tusa RJ et al: 'Joubert syndrome' revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997; 12: 423–430.
Poretti A, Huisman TA, Scheer I, Boltshauser E : Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol 2011; 32: 1459–1463.
Romani M, Micalizzi A, Valente EM : Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013; 12: 894–905.
Acknowledgements
This work was supported by the European Research Council (ERC Starting Grant 260888).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Micalizzi, A., Poretti, A., Romani, M. et al. Clinical, neuroradiological and molecular characterization of cerebellar dysplasia with cysts (Poretti–Boltshauser syndrome). Eur J Hum Genet 24, 1262–1267 (2016). https://doi.org/10.1038/ejhg.2016.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.19
This article is cited by
-
Case Report: An Adult Case of Poretti-Boltshauser Syndrome Diagnosed by Medical Checkup
The Cerebellum (2024)
-
The genetic spectrum of congenital ocular motor apraxia type Cogan: an observational study, continued
Orphanet Journal of Rare Diseases (2023)
-
Rapid exome sequencing in critically ill infants: implementation in routine care from French regional hospital’s perspective
European Journal of Human Genetics (2022)
-
Diagnostic Approach to Cerebellar Hypoplasia
The Cerebellum (2021)
-
Get Your Molar Tooth Right: Joubert Syndrome Misdiagnosis Unmasked by Whole-Exome Sequencing
The Cerebellum (2021)
