
Abstract
Anterior segment dysgenesis (ASD) encompasses a broad spectrum of developmental conditions affecting anterior ocular structures and associated with an increased risk for glaucoma. Various systemic anomalies are often observed in ASD conditions such as Axenfeld-Rieger syndrome (ARS) and De Hauwere syndrome. We report DNA sequencing and copy number analysis of PITX2 and FOXC1 in 76 patients with syndromic or isolated ASD and related conditions. PITX2 mutations and deletions were found in 24 patients with dental and/or umbilical anomalies seen in all. Seven PITX2-mutant alleles were novel including c.708_730del, the most C-terminal mutation reported to date. A second case of deletion of the distant upstream but not coding region of PITX2 was identified, highlighting the importance of this recently discovered mechanism for ARS. FOXC1 deletions were observed in four cases, three of which demonstrated hearing and/or heart defects, including a patient with De Hauwere syndrome; no nucleotide mutations in FOXC1 were identified. Review of the literature identified several other patients with 6p25 deletions and features of De Hauwere syndrome. The 1.3-Mb deletion of 6p25 presented here defines the critical region for this phenotype and includes the FOXC1, FOXF2, and FOXQ1 genes. In summary, PITX2 or FOXC1 disruptions explained 63% of ARS and 6% of other ASD in our cohort; all affected patients demonstrated additional systemic defects with PITX2 mutations showing a strong association with dental and/or umbilical anomalies and FOXC1 with heart and hearing defects. FOXC1 deletion was also found to be associated with De Hauwere syndrome.
Similar content being viewed by others

Introduction
Anterior segment dysgenesis (ASD) disorders encompass a broad spectrum of conditions associated with abnormal development of structures located in the anterior segment of the eye and an increased risk for glaucoma.1, 2 One common form of ASD is Axenfeld-Rieger syndrome (ARS), characterized by specific ocular anomalies with or without systemic abnormalities. Ocular findings include posterior embryotoxon, iris malformation, corectopia/polycoria, irido-corneal adhesions, and ∼50% risk for glaucoma.3, 4 Common systemic abnormalities include craniofacial dysmorphism with maxillary hypoplasia, hypodontia, and umbilical anomalies; hearing loss, heart anomalies, developmental delay, and other variable features have also been reported.3, 5
The homeodomain-containing transcription factor PITX2 located at 4q25 was the first ARS gene to be identified.6 A second gene, the forkhead transcription factor FOXC1 located at 6p25, has also been linked to ARS.7, 8 Mutations in these two genes, PITX2 and FOXC1, are estimated to explain ∼40% of ARS.5, 9, 10 In general, mutations in the PITX2 gene appear more likely to be associated with ocular, dental, and umbilical anomalies, whereas mutations in FOXC1 appear to be associated with isolated ocular or ocular, heart, and/or hearing defects.5, 10, 11 The phenotype associated with mutations in both of these genes is variable; even within a single family there is often variation in the specific combination of features that are seen.5, 10, 11
The human PITX2 mutations identified to date cluster in the homeodomain and C-terminal region,5, 9, 10 and mainly result in a complete or partial loss of function, with mutant proteins that retain some wild-type activity producing milder phenotypes.12, 13, 14 Dominant-negative and gain-of-function mutations have also been reported but represent an apparent minority.15, 16, 17 Other types of PITX2 mutations include deletions of coding exons and chromosomal translocations.5, 6, 18 A novel mechanism, deletion of an upstream regulatory region of PITX2, was recently reported in one patient with ARS.19
Human FOXC1 mutations associated with ARS include missense mutations in the forkhead domain, nonsense and frameshift mutations throughout the gene, and whole gene deletions.5, 10 As with PITX2, these mutations primarily result in complete or partial loss of function, particularly affecting the protein's transactivational function.5 Duplications of FOXC1 have also been reported in various types of anterior segment disorders.5, 11, 20
Another condition associated with ARS is De Hauwere syndrome, characterized by anterior chamber eye defects, hypertelorism, psychomotor retardation, hypotonia, hearing loss, femoral head anomalies, and hydrocephalus/enlarged ventricles.21, 22 The first familial occurrence was described by De Hauwere et al21 in 1973. Previous screening of the PITX2-, FOXC1-, and BARX1-coding regions in a patient with De Hauwere syndrome did not identify any mutations.22
In this manuscript, we present PITX2 and FOXC1 DNA sequencing and copy number analysis of 71 patients affected with ASD (including 38 with ARS) with or without systemic defects, 4 patients with related conditions, and 1 patient with De Hauwere syndrome.
Methods
Human subjects
This human study was approved by the Institutional Review Boards of the Children's Hospital of Wisconsin and the University of Iowa. Signed informed consent was provided by all participants and/or their legal guardians, as appropriate. We present DNA sequencing and copy number analysis of PITX2 and FOXC1 in 38 probands with ARS and 33 with other ASD (primarily Peters anomaly), with or without non-ocular defects, 4 probands with overlapping non-ocular features but other eye phenotypes, and 1 proband with De Hauwere syndrome whose clinical diagnosis, features, and photographs were presented previously;22 19 of the probands had an affected family member (16 ARS, 2 other ASD, and 1 other).
Gene sequencing
The PITX2 and FOXC1 genes were analyzed by direct DNA sequencing of PCR products encompassing all coding exons and exon/intron junctions. For PITX2, sequence was obtained for PITX2A, PITX2B, and PITX2C isoforms, using the previously described primers and conditions;23 for FOXC1, FOXC1ii, FOXC1iii, and FOXC1iv primers and conditions described by Kaur et al24 were utilized in combination with the following new primer set to include the 5′ region of the gene: FOXC1-5′F, GAGACCGAGAAAAGGTGACG, and FOXC1-5′R, AAGCGGTCCATGATGAACTGG. The PCR product of 814 bp was amplified using the following conditions: initial denaturation at 98° for 5 min followed by 35 cycles of 98 (1 min), 61 (1 min), and 72°C (1 min 30 s) and then 7-min final extension at 72°C. All DNA sequences were analyzed manually and using Mutation Surveyor (SoftGenetics, State College, PA, USA). All mutations were confirmed in an independent sequencing reaction. Mutations were named according to the most commonly used reference sequences, NM_153427.1 (PITX2A) and NM_001453.2 (FOXC1). In addition, 180 Caucasian controls were screened for variation in PITX2, and 76 Caucasian and 74 African American controls were screened for variation in FOXC1; identified variations were also compared with the ∼5000 exomes included in the Exome Variant Server (http://evs.gs.washington.edu/EVS/).25
TaqMan assays and Affymetrix copy number analyses
Patients were screened as previously described using TaqMan assays (Applied Biosystems/Life Technologies, Carlsbad, CA, USA) for the PITX2 and FOXC1 regions and/or Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA);19, 26 clinical Agilent 105K oligonucleotide array (Agilent Technologies, Santa Clara, CA, USA) and Affymetrix 6.0 array data were used for one patient each (cases 21 and 27, respectively). The following PITX2 probes were used for TaqMan assays: Hs00452261_cn (P1, located in the last exon of PITX2), Hs00958157_cn (P2, PITX2C promoter), Hs01402614_cn (P3, most 5' PITX2 exon, exon 1A), Hs06705585_cn (P3B, located at ∼50 kb upstream of PITX2), Hs04822300_cn (P4, located 110 366 kb 5' of PITX2), Hs04838001_cn (P5, located 284 481 kb 5' of PITX2), and Hs04811562_cn (P6, located 649 476 upstream of PITX2). The following 6p25 region TaqMan probes were used: Hs03037749_cn, located in the middle of FOXC1 (single exon) and Hs00919636_cn, targeting exon 2 of GMDS.
Results
A total of 28 mutations in PITX2 or FOXC1 were identified in probands, explaining 37% (26/71) of all ASD (including ARS) and 63% (24/38) of ARS specifically; the final two mutations were found in one patient without ASD (1/4; 25%) and the patient with De Hauwere syndrome. Among these mutations, 18 are nucleotide changes in exons (13) or introns (5), 9 are deletions of the coding region, and 1 is a deletion of the distant upstream region. The race/ethnicity of the probands included 7 Caucasian, 2 Hispanic, 2 of mixed race, 1 other, and 16 unknown/unreported. None of the identified mutations were seen in the controls screened here or in the ∼5000 exomes included in the Exome Variant Server.25 Mutations identified in this study were submitted to Leiden Open Variation Database (https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php).
PITX2 mutations: phenotypes and genotypes
PITX2 mutations and deletions were identified in 24 unrelated cases (Table 1, Figure 1), all of whom demonstrated dental and/or umbilical anomalies. In addition, several previously reported polymorphisms were observed in both patient and control populations. Mutations/deletions were identified in 18/18 (100%) probands with ARS and dental and umbilical anomalies, 3/9 (33%) with ARS and either dental or umbilical anomalies, 2/7 (29%) with other ASD and either dental or umbilical anomalies, and 1/4 (25%) with umbilical and/or dental anomalies without ASD (Table 1). Overall, PITX2 disruption was identified in 21/38 (55%) of all ARS and 21/27 (78%) of ARS with dental and/or umbilical defects.

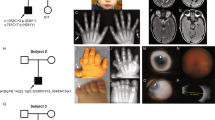
Photograph of a patient (case 11) with ARS, demonstrating the characteristic facial features (a, b), dental defects (c, d), and umbilical anomaly (e). The color reproduction of this figure is available at the European Journal of Human Genetics online.
Other less frequently observed systemic defects in probands with PITX2 abnormalities included developmental delay (5/24; 21%), growth disorder (short stature or failure to thrive) (3/24; 13%), Meckel's diverticulum (3/24; 13%), anal anomalies (2/24; 8%), heart defects (2/24; 8%), hearing loss (1/24; 4%), omphalocele (1/24; 4%), and hypospadias (1/24; 4%). Notable ocular observations were congenital glaucoma in two individuals (2/24; 8%; in contrast to the later onset developmental glaucoma usually described) and the lack of characteristic ARS ocular features in three patients (3/24; 13%). Our overall frequency of glaucoma (7/24; 29%) was lower than expected and likely reflects the young age of many participants; among patients >20-years-old, 5/9 (56%) had glaucoma. Finally, a natal tooth was reported in one patient (case 21), which is the first observation of this feature in conjunction with ARS/PITX2 mutation to our knowledge.
A total of 14 different nucleotide mutations were observed (3 mutations were seen in more than one case) (Table 1). Seven of these mutations (seen in nine cases) are novel and are consistent with previously reported mechanisms. The remaining nine cases represent new occurrences of previously reported changes.5
The previously reported c.253-11A>G mutation represented the most frequent mutation in our sample, and was associated with variation in phenotype in the three affected families (cases 7–9; Table 1). Case 7 is affected with ARS, dental, and umbilical anomalies. Case 8 has dental and umbilical anomalies along with strabismus and amblyopia, but no structural eye anomalies. Case 9 has Peters anomaly with umbilical anomalies, but no dental defects.
A novel intronic mutation c.253-1G>A, was identified in two independent families with ocular, dental, and umbilical anomalies (cases 10 and 11), and is predicted to disrupt the 3′ splice site. This is expected to lead to cryptic splice site activation or exon skipping, and to result in the production of an aberrant protein product or decreased protein production. Analysis by the CRYP-SKIP program27 (http://cryp-skip.img.cas.cz/) identifies three 3′ splice sites with a predicted strength of ≥0.5 (out of 1.0). Utilization of any one of these three sites would be predicted to cause deletion of 233, 430, or 866 nucleotides, respectively, all resulting in frameshift and truncation of the PITX2 protein.
Another novel mutation, c.708_730del (p.S237Afs*48), seen in case 18 with ocular and umbilical defects but unknown dental status (<1 year of age), is the most 3′ mutation reported to date (case 18). This mutation causes frameshift/truncation and was not seen in the180 controls screened here or in the ∼5000 exomes included in the Exome Variant Server.25 As this mutation is located in the final exon of the gene, it is not expected to be subject to nonsense-mediated decay,28 suggesting that disruption of the 14-amino-acid motif is sufficient to impair gene function.
Genomic analysis identified deletions involving the PITX2 coding region in five patients and a deletion involving the distant upstream region of PITX2 in one patient, which was located at least 50 kb upstream of PITX2 and did not affect any PITX2 exons (Table 1, Figure 2). The extent of the deletion was characterized by array data in three cases (cases 19–21) and ranged in size from at least 1.1 Mb to 19.2 Mb, deleting between 4 and 65 genes. Three cases (cases 22–24) were analyzed by PITX2-specific TaqMan assays only because of insufficient amounts of available DNA, and therefore the exact size of the deletion could not be determined.

Summary of PITX2 mutations identified in this study. (a) Schematic drawing of PITX2 gene and upstream region. Identified point mutations are shown with red numbered arrows (1–18) whereas identified deletions are shown as red lines (19–24); the solid line shows the minimum deletion size, with the dashed line representing the maximum extent of the deletions. PITX2 exons are shown as numbered boxes, regions corresponding to the homeobox and 14-amino-acid-conserved domains are indicated as black and light gray boxes, respectively. Initiation codons corresponding to the PITX2 isoforms are shown with black arrows. The stop codon is indicated with a black arrow and asterisk. The positions of the TaqMan probes are shown and labeled P1–P6; for upstream probes P3B and P4–P6, the distance (in base pairs) from the beginning of the gene is indicated in parenthesis. The positions of probes that demonstrated haploid state are shown as black diamonds, whereas probes that showed diploid state are shown as gray diamonds. (b) DNA sequencing chromatograms for normal and mutants alleles; mutation position is indicated with an arrow. (c) Copy number analysis (TaqMan assay and Affymetrix array) data. For PITX2 TaqMan assays, probes P1–P3 correspond to PITX2 exons and probes P3B and P4–P6 correspond to the upstream region, patient sample values were compared with control (C) results for every assay. Probes that showed half of the normal value are noted in red. For Affymetrix array analysis, Genotyping Console representation of chromosome 4 marker data is shown with heterozygous deletions indicated with arrows. The color reproduction of this figure is available at the European Journal of Human Genetics online.
Family members were available for testing for 13 cases. Unaffected family members were available for cases 2, 4, 6, 7, 9, 10, 11, 15, 18, 21, 22, 23, and 24; none of these family members carried the mutation/deletion identified in the proband. The mother of case 24 (with upstream deletion) has isolated posterior embryotoxon, but she did not show evidence of the deletion seen in her son. Affected family members were available for two families (cases 2B, 2C, and 4B), all of whom carried the same mutation seen in the proband (Table 1).
FOXC1 mutations: phenotypes and genotypes
No intragenic mutations in the FOXC1 gene were identified. Several previously reported polymorphisms were observed in both patient and control populations. Genomic analysis identified deletions involving the FOXC1-coding region in three probands with ARS (cases 25–27) as well as the case of De Hauwere syndrome (case 28)22 (Table 2, Figure 3). Deletion size ranged from at least 0.98 Mb to at least 1.5 Mb, deleting between 2 and 11 genes. For the three probands with ARS (cases 25–27), the deletion included both FOXC1 and GMDS (cases 25–27) with nine additional 6p25.2 genes deleted in one case (case 27). The telomeric deletion break point was positioned 18–40 kb from FOXC1. Therefore FOXF2, the next gene telomeric to FOXC1, was not included in any of these deletions with a distance of 175–197 kb between the deletion start and the FOXF2 gene. For the patient with De Hauwere syndrome (case 28), the telomeric deletion break point was localized 717 kb from FOXC1, and therefore included both the FOXF2 and FOXQ1 genes in addition to FOXC1 and GMDS (Figure 3). This patient was also found to carry a 0.52-Mb duplication of 6p21.33–p21.33 including HLA-A, HCG9, ZNRD1, PPP1R11, RNF39, TRIM31, TRIM40, TRIM10, TRIM15, TRIM26, FLJ45422, TRIM39, and RPP21. Three of the four probands with FOXC1 deletion (cases 25, 26A, and 28) demonstrated heart or hearing defects, and three (cases 25, 26A, and 27) had glaucoma (congenital/juvenile onset). All had normal dentition and umbilicus; one family member demonstrated mildly redundant periumbilical skin (case 26B). Family members were available for testing in one case; TaqMan assay in case 26B (the affected child of case 26A) identified deletion of FOXC1, as seen in his mother; Affymetrix array data was not available to determine the extent of the deletion in the child.

Summary of FOXC1 deletions identified in this study. (a) Schematic drawing of the 6p25 region. All known human protein-coding and non-protein-coding genes from the NCBI RNA reference sequences collection (RefSeq) for the selected region are shown as seen in the UCSC Genome Browser (http://www.genome/ucsc.edu); GRCh37/hg19 assembly was utilized. Identified deletions are shown as red lines (25–28); the solid line shows the minimum deletion size, with the dashed line representing the maximum extent of the deletions. The positions of the TaqMan probes that demonstrated haploid state are shown as black diamonds; the positions of the Affymetrix probes that exhibited haploid state are shown as black circles and probes that displayed diploid state as gray circles. (b) Affymetrix array identification of FOXC1/6p25 deletions. Genotyping Console representation of chromosome 6 marker data for cases 25, 26 and 28 is shown with FOXC1 position within the heterozygous deletions indicated with arrows. (c) Copy number analysis using TaqMan assays showing deletion of FOXC1 and GMDS for cases 25, 26 and 28. Patient sample values were compared with control (C) results for every assay. The color reproduction of this figure is available at the European Journal of Human Genetics online.
Discussion
PITX2 or FOXC1 disruption was found to explain 63% (24/38) of ARS specifically but only 6% (2/33) of other ASD (non-ARS), yielding 37% (26/71) overall mutation frequency in our ASD cohort. Consistent with other reports, our data provide strong evidence that PITX2 disruption is associated with ARS with dental and/or umbilical anomalies whereas FOXC1 alterations are primarily seen in ARS with isolated ocular or ARS with heart and/or hearing defects.5, 11 This suggests that despite the significant overlap in ocular phenotypes, non-ocular features may provide distinction between the syndromes associated with these two genes. In addition, 6p25 deletion was identified in a patient with De Hauwere syndrome and a minimal ∼1.3 Mb deletion region was shown to be strongly associated with this phenotype.
PITX2 mutations alone were found to explain 55% of ARS specifically (78% of ARS with dental and/or umbilical defects) and 6% of other ASD (non-ARS), yielding 33% overall PITX2 mutation frequency in our ASD population. Two previous studies that screened for both nucleotide mutations and genomic deletions of PITX2 reported a lower proportion of cases explained by disruption of PITX2: 4.7%18 and 16%10 of ASD (neither study provided the proportion of ARS specifically). The variation is most likely due to differences in sample composition as our cohort is enriched with ARS cases with additional systemic defects, particularly dental and umbilical anomalies that appear to be strongly associated with PITX2 mutations. In the study reported by Dr D'haene et al,10 11 out of 13 families with PITX2 mutations had both dental and umbilical defects (six with ARS, three with other ASD, and two with no data on ocular features), one family demonstrated ARS and dental anomalies whereas the umbilicus was not examined and no records were available for the last PITX2-positive family; heart and hearing defects were each seen in one family with PITX2 mutation.10 Another interesting phenotypic observation is that three patients with intragenic PITX2 mutations in our study were found to display a growth disorder (13%). This finding provides further support for a potential role of PITX2 in SHORT syndrome and similar conditions.18, 29
Of particular note are three PITX2 mutations reported in our study. First, the deletion we identified involving the distant upstream region (but not the coding region) of PITX2 in one ARS patient is similar to our previously reported ARS case,19 and thus our report highlights the importance of this recently discovered mechanism, loss of the upstream regulatory region, as a cause of ARS. It will be crucial to screen for this type of mutation in additional patients with ARS in order to determine its frequency. Disease-causing mutations outside of the coding region have been identified in other genes; for example, deletion of the downstream regulatory region of PAX6 was found in a number of families affected with aniridia.2, 30 Disruption of the regulatory regions of genes should be considered as a mechanism for other conditions not fully explained by intragenic mutations.
Second, the novel c.708_730del (p.S237Afs*48) mutation represents the most C-terminal alteration identified to date. This mutation occurs in the middle of a 14- amino-acid-conserved domain of yet unknown function6 providing further support for the functional significance of this motif. Finally, the c.253-11 A>G mutation in PITX2 in three of our cases appears to represent a ‘hot spot’ as it has now been reported in six independent families (cases 7–9, our study;6, 10, 31). Two of our three probands carrying this mutation have atypical ocular phenotypes. This mutation was studied extensively by Maciolek et al14 and was shown to shift splicing exclusively to a new AG acceptor site created 11 nucleotide upstream. The resulting PITX2 protein is truncated at 138 residues followed by 117 frame-shifted amino acids.14 Owing to its distance from the splice site, this mutation may be easily missed in routine genetic screening.
Deletion of FOXC1 was identified in four probands. No intragenic mutations were detected. The frequency of FOXC1 deletion in ARS was 8% (3/38), which is lower than previously reported. D'haene et al10 found that both mutations and deletions involving FOXC1 accounted for 24% of patients with ARS or other ASD with and without non-ocular anomalies. The difference between their study and ours is most likely because of bias toward ARS with dental and umbilical defects in our cohort, and the higher proportion of PITX2 mutations in those phenotypes. Three individuals with FOXC1 deletion in our study demonstrated hearing loss and two had heart abnormalities; these results are consistent with previous reports of FOXC1 mutations.5, 10 Interestingly, a higher proportion of FOXC1 probands were affected with glaucoma (3/4; 75%) compared with PITX2 probands (7/24; 29%). A similar increase in glaucoma was seen in a previous study with 12/16 (75%) FOXC1 cases compared with 5/12 (42%) PITX2 cases affected with glaucoma.10 It is not clear whether FOXC1 disruption is truly associated with an increased risk of glaucoma or perhaps an earlier onset.
The FOXC1 deletions identified in the three ARS probands (cases 25–27) in our study include one or more genes centromeric to FOXC1, which is consistent with the previous reports of patients with ARS.10, 20 Of special interest is the deletion identified in the patient with De Hauwere syndrome, which is the only one of the deletions reported here that extends in the telomeric direction. De Hauwere syndrome (OMIM: 109120), characterized by anterior segment eye defects, hypertelorism, psychomotor retardation, hypotonia, hearing loss, hydrocephalus/enlarged ventricles, and femoral head anomalies,21, 22 displays a significant overlap with 6p25 deletion syndrome (OMIM: 612582), defined as a combination of ocular anomalies (primarily anterior segment), hearing loss, congenital heart disease, hydrocephalus, developmental delay, and a characteristic facial appearance and typically associated with terminal deletions of 6p25.32, 33 Review of the literature identified that the skeletal features observed in De Hauwere syndrome, such as flattening of the femoral epiphyses and other femoral head anomalies sometimes diagnosed as Perthes disease, were reported in several previously described patients with 6p25 terminal deletions (Table 3),32, 34, 35, 36, 37, 38 suggesting that De Hauwere syndrome may be part of the 6p25 deletion syndrome spectrum. Case 28 in our study has the smallest and first interstitial deletion reported to date in association with the features of De Hauwere syndrome, thus defining a minimal deleted region for this phenotype.
The four genes affected in De Hauwere syndrome (case 28, this study), FOXQ1, FOXF2, FOXC1, and GMDS, are also deleted in the six previously described patients with features of De Hauwere syndrome (Table 3). A minimal deleted region in the 6p25 deletion syndrome was previously defined as ∼2.1 Mb from the telomere to FOXC1, thus including FOXQ1 and FOXF2 but not GMDS.39 GMDS is deleted in our other ARS cases and numerous other patients with interstitial 6p25 deletions lacking skeletal/other anomalies characteristic of De Hauwere syndrome (Table 2,10), suggesting that deletion of GMDS is not critical to the De Hauwere phenotype. The remaining deleted genes associated with features of De Hauwere syndrome, FOXC1, FOXF2, and FOXQ1, have been shown to have a role in the skeletal or craniofacial cartilage development in animal models. Mice homozygous for a null allele of the FOXC1 homolog Mf1 died shortly after birth and displayed an ASD, hydrocephalus, craniofacial defects, and skeletal abnormalities, including small, malformed, or missing bones and reduced ossification centers in the appendages;40, 41 evaluation of heterozygous mice revealed variable anterior segment ocular defects and some mild delays in ossification or reduction in the ossification centers of the sternum.40 Mice homozygous for a Foxf2 knockout allele exhibited cleft of the secondary palate and died within 12–18 h of birth with other defects secondary to the cleft and no other skeletal anomalies;42 a recent study of a Foxf2 missense mutation suggested that the Foxf2 gene also has a role in anterior segment ocular development in mice, whereas palatal defects were not observed.43 Although no skeletal defects were reported in Foxq1 deficient mice,44 one recent study detected Foxq1 expression in the developing craniofacial cartilage in zebrafish.45 Further studies of patients with terminal and interstitial deletions will help to define which genes are critical for different aspects of 6p25 deletion phenotypes.
In summary, mutations in PITX2 and FOXC1 explain a significant portion of ARS phenotypes. Non-ocular features can be efficiently utilized to guide genetic testing strategy as dental and umbilical anomalies are strongly associated with PITX2, whereas heart and hearing defects show association with FOXC1. Identification of an interstitial 6p25 deletion encompassing FOXC1 and other forkhead genes in the 6p25 region in a patient with De Hauwere syndrome, together with the previously reported cases, provides strong evidence for involvement of FOXC1 in this condition and defines the likely critical interval.
References
Sowden JC : Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond) 2007; 21: 1310–1318.
Reis LM, Semina EV : Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol 2011; 22: 314–324.
Rieger H : Verlagerung und schlitzform der pupille mit hypoplasie des irisvorderblattes. Z Augenheilkd 1934; 84: 98–103.
Alward WL : Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol 2000; 130: 107–115.
Tümer Z, Bach-Holm D : Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet 2009; 17: 1527–1539.
Semina EV, Reiter R, Leysens NJ et al: Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996; 14: 392–399.
Mears AJ, Jordan T, Mirzayans F et al: Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet 1998; 63: 1316–1328.
Nishimura DY, Swiderski RE, Alward WL et al: The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet 1998; 19: 140–147.
Hjalt TA, Semina EV : Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med 2005; 7: 1–17.
D'haene B, Meire F, Claerhout I et al: Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Invest Ophthalmol Vis Sci 2011; 52: 324–333.
Strungaru MH, Dinu I, Walter MA : Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci 2007; 48: 228–237.
Kozlowski K, Walter MA : Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum Mol Genet 2000; 9: 2131–2139.
Espinoza HM, Cox CJ, Semina EV, Amendt BA : A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum Mol Genet 2002; 11: 743–753.
Maciolek NL, Alward WL, Murray JC, Semina EV, McNally MT : Analysis of RNA splicing defects in PITX2 mutants supports a gene dosage model of Axenfeld-Rieger syndrome. BMC Med Genet 2006; 7: 59.
Priston M, Kozlowski K, Gill D et al: Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum Mol Genet 2001; 10: 1631–1638.
Saadi I, Semina EV, Amendt BA et al: Identification of a dominant negative homeodomain mutation in Rieger syndrome. J Biol Chem 2001; 276: 23034–23041.
Saadi I, Toro R, Kuburas A, Semina E, Murray JC, Russo AF : An unusual class of PITX2 mutations in Axenfeld-Rieger syndrome. Birth Defects Res A Clin Mol Teratol 2006; 76: 175–181.
Lines MA, Kozlowski K, Kulak SC et al: Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci 2004; 45: 828–833.
Volkmann BA, Zinkevich NS, Mustonen A et al: Potential novel mechanism for axenfeld-rieger syndrome: deletion of a distant region containing regulatory elements of PITX2. Invest Ophthalmol Vis Sci 2011; 52: 1450–1459.
Lehmann OJ, Ebenezer ND, Ekong R et al: Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Invest Ophthalmol Vis Sci 2002; 43: 1843–1849.
De Hauwere RC, Leroy JG, Adriaenssens K, Van Heule R : Iris dysplasia, orbital hypertelorism, and psychomotor retardation: a dominantly inherited developmental syndrome. J Pediatr 1973; 82: 679–681.
Lowry RB, Gould DB, Walter MA, Savage PR : Absence of PITX2, BARX1, and FOXC1 mutations in De Hauwere syndrome (Axenfeld-Rieger anomaly, hydrocephaly, hearing loss): a 25-year follow up. Am J Med Genet A 2007; 143A: 1227–1230.
Bremond-Gignac D, Bitoun P, Reis LM, Copin H, Murray JC, Semina EV : Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol Vis 2010; 16: 1705–1711.
Kaur K, Ragge NK, Ragoussis J : Molecular analysis of FOXC1 in subjects presenting with severe developmental eye anomalies. Mol.Vis 2009; 15: 1366–1373.
Exome Variant Server: NHLBI Exome Sequencing Project (ESP). Seattle, WA, November 2011.
Reis LM, Tyler RC, Schilter KF et al: BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum Genet 2011; 130: 495–504.
Divina P, Kvitkovicova A, Buratti E, Vorechovsky I : Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur J Hum Genet 2009; 17: 759–765.
Nagy E, Maquat LE : A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998; 23: 198–199.
Karadeniz NN, Kocak-Midillioglu I, Erdogan D, Bokesoy I : Is SHORT syndrome another phenotypic variation of PITX2? Am J Med Genet A 2004; 130A: 406–409.
Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T : 3' deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci USA 2000; 97: 13755–13759.
Borges AS, Susanna R, Carani JC et al: Genetic analysis of PITX2 and FOXC1 in Rieger Syndrome patients from Brazil. J Glaucoma 2002; 11: 51–56.
Gould DB, Jaafar MS, Addison MK et al: Phenotypic and molecular assessment of seven patients with 6p25 deletion syndrome: relevance to ocular dysgenesis and hearing impairment. BMC Med Genet 2004; 5: 17.
DeScipio C : The 6p subtelomere deletion syndrome. Am J Med Genet C Semin Med Genet 2007; 145C: 377–382.
Mirza G, Williams RR, Mohammed S et al: Refined genotype-phenotype correlations in cases of chromosome 6p deletion syndromes. Eur J Hum Genet 2004; 12: 718–728.
Maclean K, Smith J, Heaps L et al: Axenfeld-Rieger malformation and distinctive facial features: clues to a recognizable 6p25 microdeletion syndrome. Am J Med Genet A 2005; 132: 381–385.
Kannu P, Oei P, Slater HR, Khammy O, Aftimos S : Epiphyseal dysplasia and other skeletal anomalies in a patient with the 6p25 microdeletion syndrome. Am J Med Genet A 2006; 140: 1955–1959.
Martinez-Glez V, Lorda-Sanchez I, Ramirez JM et al: Clinical presentation of a variant of Axenfeld-Rieger syndrome associated with subtelomeric 6p deletion. Eur J Med Genet 2007; 50: 120–127.
Bedoyan JK, Lesperance MM, Ackley T, Iyer RK, Innis JW, Misra VK : A complex 6p25 rearrangement in a child with multiple epiphyseal dysplasia. Am J Med Genet A 2011; 155A: 154–163.
Anderlid BM, Schoumans J, Hallqvist A et al: Cryptic subtelomeric 6p deletion in a girl with congenital malformations and severe language impairment. Eur J Hum Genet 2003; 11: 89–92.
Hong HK, Lass JH, Chakravarti A : Pleiotropic skeletal and ocular phenotypes of the mouse mutation congenital hydrocephalus (ch/Mf1) arise from a winged helix/forkhead transcriptionfactor gene. Hum Mol Genet 1999; 8: 625–637.
Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL : The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 1998; 93: 985–996.
Wang T, Tamakoshi T, Uezato T et al: Forkhead transcription factor Foxf2 (LUN)-deficient mice exhibit abnormal development of secondary palate. Dev Biol 2003; 259: 83–94.
McKeone R, Vieira H, Gregory-Evans K, Gregory-Evans CY, Denny P : Foxf2: a novel locus for anterior segment dysgenesis adjacent to the foxc1 gene. PLoS One 2011; 6: e25489.
Hong HK, Noveroske JK, Headon DJ et al: The winged helix/forkhead transcription factor Foxq1 regulates differentiation of hair in satin mice. Genesis 2001; 29: 163–171.
Planchart A, Mattingly CJ : 2,3,7,8-Tetrachlorodibenzo-p-dioxin upregulates FoxQ1b in zebrafish jaw primordium. Chem Res Toxicol 2010; 23: 480–487.
Acknowledgements
We thank the patients and their families for their participation in research studies. We are also thankful to Drs William Rhead, Peter Francis, Omar Abdul-Rahman, and Annet Van Hagen for referral of patients; Jennifer Rigdon, Kate Durda, Jamie L'Heureux, and Ecaterina Dragan for assistance with sample processing and clinical data collection; and Rachel Lorier, Stephen Hall, Katie Felhofer and Andrea Lenarduzzie for assistance with Affymetrix array CNV analysis. This work was supported by the National Institutes of Health awards EY015518 (EVS), 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) Program, Research Training Program in Vision Sciences T32 EY014537, and the Foerderer Fund (AVL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Reis, L., Tyler, R., Volkmann Kloss, B. et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet 20, 1224–1233 (2012). https://doi.org/10.1038/ejhg.2012.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.80
Keywords
This article is cited by
-
CRISPR-Cas9-mediated functional dissection of the foxc1 genomic region in zebrafish identifies critical conserved cis-regulatory elements
Human Genomics (2022)
-
A novel missense mutation of FOXC1 in an Axenfeld–Rieger syndrome patient with a congenital atrial septal defect and sublingual cyst: a case report and literature review
BMC Medical Genomics (2021)
-
A novel variant in FOXC1 associated with atypical Axenfeld-Rieger syndrome
BMC Medical Genomics (2021)
-
A missense mutation in Pitx2 leads to early-onset glaucoma via NRF2-YAP1 axis
Cell Death & Disease (2021)
-
Pitx genes in development and disease
Cellular and Molecular Life Sciences (2021)
