
Abstract
Mutations in the four-and-a-half LIM domain 1 (FHL1) gene, which encodes a 280-amino-acid protein containing four LIM domains and a single zinc-finger domain in the N-terminal region, have been associated with a broad clinical spectrum of X-linked muscle diseases encompassing a variety of different phenotypes. Patients might present with a scapuloperoneal myopathy, a myopathy with postural muscle atrophy and generalized hypertrophy, an Emery–Dreifuss muscular dystrophy, or an early onset myopathy with reducing bodies. It has been proposed that the phenotypic variability is related to the position of the mutation within the FHL1 gene. Here, we report on three British families with a heterogeneous clinical presentation segregating a single FHL1 gene mutation and haplotype, suggesting that this represents a founder mutation. The underlying FHL1 gene mutation was detected by direct sequencing and the founder effect was verified by haplotype analysis of the FHL1 gene locus. A 3-bp insertion mutation (p.Phe127_Thr128insIle) within the second LIM domain of the FHL1 gene was identified in all available affected family members of the three families. Haplotype analysis of the FHL1 region on Xq26 revealed that the families shared a common haplotype. The p.Phe127_Thr128insIle mutation in the FHL1 gene therefore appears to be a British founder mutation and FHL1 gene screening, in particular of exon 6, should therefore be indicated in British patients with a broad phenotypic spectrum of X-linked muscle diseases.
Similar content being viewed by others


Introduction
The human four-and-a-half LIM domain 1 (FHL1) gene, located on Xq26.3, encodes for a 32-kDa protein with four and a half tandem repeat LIM domains. LIM domains are cysteine-rich, tandem zinc-finger protein interaction motifs first recognized in the three homeodomain transcription factors Lin-11, Isl-1 and Mac-3.1, 2, 3, 4, 5 FHL1 has at least three isoforms, a full-length protein (FHL1A) and two additional variants referred to as FHL1B and C, differing in their primary structure, expression pattern, binding partners and subcellular localizations.6, 7 All isoforms are highly expressed in skeletal muscle and to a lesser extent also in heart muscle (left and right ventricle).8 FHL1 localizes to the sarcomere and the sarcolemma and is believed to participate in muscle growth and differentiation as well as in sarcomere assembly.8
We first described FHL1 gene mutations in families presenting with an X-linked myopathy with postural muscle atrophy and generalized hypertrophy (XMPMA).9 Concurrently, FHL1 gene mutations have been detected in X-linked dominant scapuloperoneal myopathy,10, 11 and subsequently in a wide spectrum of partly overlapping conditions, such as reducing body myopathy (RBM),12, 13, 14 rigid spine syndrome,15 Emery–Dreifuss muscular dystrophy (EDMD),16 and in a single family with contractures, rigid spine, and cardiomyopathy.17 It has been suggested to classify FHL1-related disorders into two main groups, the ‘reducing body (RB) subgroup’ including RBM, X-linked dominant scapuloperoneal myopathy and rigid spine syndrome, all characterized by RB at histological examination, and a second subgroup, including the XMPMA and EDMD patients, characterized by later onset and absence of RB.12, 13, 16 RBM may also be seen as a condition within a larger phenotypic spectrum of protein aggregate myopathies, such as the myofibrillar myopathies.18
Here, we report three British families with X-linked muscle diseases, one being part of our original report,9 presenting with partly heterogeneous phenotypes but sharing the p.Phe127_Tyr128insIle mutation in the FHL1 gene and a common Xq26 haplotype.
Subjects and methods
Clinical assessment
Three families were included in the study. Their case histories were reviewed and seven patients were interviewed and examined. Their age ranged between 27 and 55 years at the time of the study. Respiratory function was assessed clinically and spirometrically. In six patients, a formal cardiology assessment was also performed. Clinical information for 13 additional now-deceased family members was provided by the patients and their families. A muscle biopsy was obtained for diagnostic purposes from at least one individual in each of the families. All patients included in this study provided appropriate consent for the investigations reported.
Muscle biopsy
Skeletal muscle biopsies were assessed by routine histochemistry and immunoanalysis. Optimised immunohistochemical and multiplex western blot analysis were performed as previously described.19, 20 In addition, antibodies against myotilin (Novocastra, Newcastle upon Tyne, UK) and desmin (in-house DY14/5H2) were also used. At the time of the present study, muscle tissues and sections were not available for histological re-evaluation and menadione–NBT staining, and for FHL1 protein immunoanalysis.
Genetic analysis
Genomic DNA was extracted from blood samples using standard procedures. FHL1 mutation screening was performed using primers, amplification conditions and a protocol previously reported.9 For haplotype analysis of the FHL1 locus, the following markers were selected and analysed as previously described: DXS8009, AFMa288xd5, AFM205wd2, AFMb340zd9, DXS8074, DXS8033, DXS8094, DXS1041 and DXS1227.9
Results
Phenotypic presentation
Main clinical features of the three families are summarized in Table 1.
Family 1. Four male individuals in three consecutive generations were affected (Figure 1a, Table 1). This family has previously been reported as the ‘UK family’ in the original report by Windpassinger et al.9 Patient F1/18 showed first symptoms in his late 30s when he started noticing shoulder, proximal arm and hip muscle weakness. Weakness progressed over time and caused difficulties in walking, with myalgia in calves and thighs. The patient started using a wheelchair in his early 50s. At 55 years of age, he presented with symmetrical scapular winging and spinal rigidity, which was most pronounced in the cervical region (Figures 2a–c). On manual muscle testing, he showed profound weakness in his shoulder girdle and upper arm muscles, with the biceps and brachioradialis muscles being most severely affected (MRC power grade 1–2). He also showed prominent pelvic girdle weakness with hip adduction and abduction being severely compromised (MRC power grade 1–2). Muscles of the lower legs and feet were less severely affected (MRC power grade 3–4). Lung function was moderately reduced with a forced vital capacity of 2.43 l (63% of the predicted value) in sitting and 2.11 l (55% of the predicted value) in lying. CK values were elevated up to 588 U/l (upper normal limit 150 U/l). Patient F1/23, maternal first cousin once removed of patient F1/18, presented first symptoms of mild limb girdle weakness at 35 years of age. At that time, his overall motor performance, speed and endurance were very good and he played cricket and football without complaints, such as myalgia or other symptoms. On examination at 37 years of age, he presented with a mild waddling gait, prominent shoulder girdle and arm muscle hypertrophy, as well as mild calf asymmetry (Figure 2d). His left Achilles tendon was tight. Cervical spine rigidity was also present. Mild muscle weakness was evident in his hip flexors, abductors and adductors, and he showed neck extension weakness (MRC power grade 4). At 39 years of age, he started to experience increased gait and postural problems, as well as occasional falls and lower back pain. However, no deterioration of muscle strength and lung function was detected. His forced vital capacity showed a 13% drop from sitting to lying. ECG analysis was normal. His serum CK levels were elevated up to 1300 U/l. Two additional male relatives were reported as affected and had died. Patient F1/05 started showing motor problems in his early 40s and became wheelchair bound within 10 years after the onset of symptoms. The patient died of unknown cause at 52 years of age. Patient F1/16 showed a clinical picture similar to patient F1/15 and died in his early 50s. Respiratory failure was reported for both of them, but no information was available on their cardiac function. There was no report in the family about affected female carriers.

Pedigrees of the three families segregating the p.Phe127_Thr128insIle mutation in the FHL1 gene. Pedigrees in (a–c) represent family F1–F3, respectively. Family F1 has also been reported as the ‘UK family’ in the original report by Windpassinger et al.9 Filled symbol: affected. Empty symbol: unaffected. Dotted symbol: obligate carrier. The index patient of each family is indicated with an arrow. ‘+’ sign: individuals harbouring the FHL1 gene mutation. ‘−’ sign: females not carrying the FHL1 mutation.

Clinical assessment of two patients from family F1 with the p.Phe127_Thr128insIle mutation in the FHL1 gene. (a) Frontal, (b) lateral and (c) posterior view of patient F1/18 showing marked cervical spinal rigidity, shoulder girdle weakness and scapular winging. (d) Frontal view of patient F1/23 showing relative hypertrophy of upper limb muscles.
Family 2. Five females and six males, spread over five generations were affected (Figure 1b, Table 1). The three evaluated male subjects (F2/47, F2/49 and F2/55) showed an onset of symptoms in their second–third decade, with predominant progressive limb girdle weakness and scapular winging. The shoulder girdle and upper arm muscles were more severely affected than the muscles of the lower extremities. They also showed severe biceps weakness, cervical spinal rigidity and Achilles tendon contractures. Patients F2/47 and F2/55 are still ambulant, whereas patient F2/49 became wheelchair bound at 30 years of age. Lung function was reduced in patient F2/47 (forced vital capacity of 2.3 l, which corresponded to 52% of the predicted value) and F2/49, who showed a drop in forced vital capacity from 2.73 l in sitting (61% of the predicted value) at 32 years of age to 0.9 l (20% of the predicted value) at 38 years of age. Serum CK levels were elevated up to 1500–2200 U/l. An additional three deceased male family members from the preceding generations were reported as wheelchair bound from their 30s (F2/18, F2/26, F2/35). According to family members, all of them died in their late 40s–early 50s due to cardiorespiratory failure (Table 1). Female mutation carriers generally presented with clinical symptoms of muscle weakness in the middle age or later in life (F2/42, F2/46). They showed mild shoulder girdle weakness and distal lower limb involvement with ankle dorsiflexion weakness (Table 1). The only serum CK level available for a single female patient was mildly elevated (277 U/l, patient F2/46; Table 1). An additional three females in the family (F2/10, F2/13, F2/21) were reported to be affected by a clinical picture similar to the one we observed in the examined female patients. All of these female patients had already died at the time of the study; one of them (F2/21) at 88 years of age due to congestive heart failure (Table 1).
Family 3. Five males, spread over four generations, were reported as affected by a muscle disease (Figure 1c, Table 1). The index patient (F3/46) showed a disease onset at around 30 years of age, with proximal upper and lower limb weakness, foot drop and restricted neck movements. On examination, at 45 years of age, the patient was still ambulant, but experienced frequent falls and was unable to walk unaided. He showed severe neck rigidity and periscapular wasting. Manual muscle testing revealed severe biceps and mild lower arm weakness. His lateral thigh muscles (vastus lateralis) appeared hypertrophic, whereas medial thigh and hamstring muscles were wasted. Respiratory and cardiac function was preserved. Serum CK values were elevated between 750 and 1500 IU/l. The maternal uncle of the index patient (F3/29) died at 55 years of age and was reported to be similarly affected. Three additional affected males (F3/9, F3/13, F3/16 and F3/36; Figure 1c) in the preceding generations were described as having gait problems, but no further information on their clinical presentation was available.
Muscle histopathology
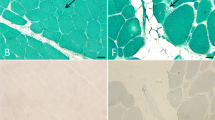
A muscle biopsy performed on patient F1/18 at 41 years of age showed a few atrophic fibres, internal nuclei in about 20% of the fibres and several ring fibres (data not shown). Immunoanalysis of muscle sections was normal. A vastus lateralis muscle biopsy from patient F1/23 performed at 34 years of age showed variation in fibre size, some internal nuclei and a few necrotic fibres (Figure 3a). Muscle histology of the left deltoid muscle from patient F2/47 at 30 years of age showed a nonspecific myopathic pattern with mild excess of type 1 fibres. Muscle immunoanalysis did not show any abnormalities (data not shown). A left-biceps biopsy from F3/46 showed fibre size variation, inflammation associated with necrotic fibres and occasional fibres containing internal nuclei (Figure 3b). A few degenerating fibres with rimmed vacuoles and eosinophilic inclusions were also noted. Immunohistochemistry, including staining for desmin and myotilin, was normal (data not shown).

Histological findings in patients with the p.Phe127_Thr128insIle mutation in the FHL1 gene. (a) H and E staining from a muscle biopsy section from patient F1/23 showing variation in fibre size, internal nuclei, a few rounded almost hyaline fibres and a few necrotic fibres. (b) H and E staining from a muscle biopsy section from patient F3/46 showing fibre size variation, inflammation associated with necrotic fibres, slight increase in fat and connective tissue, and occasional fibres containing internally located nuclei.
Clinical investigations
Nerve conduction studies were normal in patients F1/23 and F2/47. EMG studies in patient F1/23 were reported as normal, whereas EMG studies in F2/49 and F3/46 showed myopathic changes (Table 1). Cardiac investigations did not show any abnormalities in the six patients examined (patients F1/23, F2/42, F2/46, F2/47, F2/55 and F3/46).
Molecular analysis
Direct sequencing of the complete coding sequence of the FHL1 gene identified the same small in-frame insertion in exon 6 (c.381_382insATC) in all three index patients.9 This change gives rise to the insertion of an additional isoleucine within the second LIM domain (p.Phe127_Thr128insIle). The mutation was present in all the tested male family members and in a heterozygous state in two affected females of family F2. Two further unaffected females from family F2 were tested and showed no FHL1 gene change (F2/54 and F2/57). Linkage analysis for the FHL1 gene locus revealed a common haplotype spanning at least 10 Mb in the three index patients as well as in four members of family F2 (Figure 4).

Ideogrammatic representation of the FHL1 locus on the distal arm of chromosome X and haplotype analysis in one member of each family for the FHL1 gene locus on Xq26. Position of STR markers are indicated in megabases and referred to the UCSC hg19 assembly (http://genome.ucsc.edu/cgi-bin/hgGateway).
Discussion
We identified three British families segregating an identical FHL1 gene mutation and haplotype. The phenotype of the patients was characterized by a number of common clinical features, but with heterogeneous disease onset, muscle involvement and disease progression in the three families. Overall, affected individuals showed a progressive myopathy characterized by adult onset, proximal lower limb weakness with severe biceps involvement, cervical rigidity and raised CK values. Conversely, the age of disease onset was heterogeneous among affected males both within the same family and in between families, ranging from late teens in family F2 to mid 30s and 40s in families F3 and F1. The course of the disease was slowly progressive in two families (F1 and F3), and examined patients (such as patient F3/18) were still ambulant after a disease duration of 10–15 years. Conversely, in family F2, the disease had an overall more severe progression in males, with earlier onset and more rapid progression with patient F2/49 being wheelchair bound from his early 30s. Affected carrier females were observed only in family F2 and showed a mild scapuloperoneal phenotype with onset in middle age or later. Generalized muscle hypertrophy was observed in a single patient (F1/23), whereas patient F3/46 showed hypertrophy of a single muscle. Slowly progressive respiratory muscle involvement, with onset from the third–fourth decade, was observed in affected individuals from families F1 and F2, whereas cardiac disease was not observed in any of the examined individuals. Although cardiac involvement was reported for the deceased individuals from family F2, the absence of any heart involvement in the examined patients from the three families would indicate that this may not be a feature commonly associated with this FHL1 genotype or that cardiac pathology may only develop at later stages of the disease. Overall, we observed intra- and interfamilial heterogeneity with regard to age at onset of symptoms, pattern and severity of muscle involvement and disease progression.
Because of the heterogeneous phenotypes, classification of our families within one of the FHL1-related diseases appears challenging and might create confusion. From a nosological point of view, there is clear overlap between the phenotype observed in our families and the second FHL1-related disease subgroup, that is, XMPMA and EDMD, in particular because of the overall late onset clinical presentation. However, several important differences were noticeable, suggesting that the p.Phe127_Thr128InsIle FHL1 mutation might be associated with a phenotypic spectrum ranging from XMPMA as described in the original patient (F1/23) to an X-linked dominant scapuloperoneal myopathy (as in the female carriers from F2) or a more contractural phenotype, such as in patient P2/55 and P3/46. The differential diagnosis for FHL1-associated muscle diseases includes other contractural myopathies, such as Bethlem myopathy and also the myofibrillar myopathies. Once a dystrophinopathy has been excluded, increased CK values in male subjects with possible X-linked inheritance should always trigger FHL1 gene screening.
To the best of our knowledge, 26 different FHL1 mutations have been reported so far in 36 unrelated individuals affected by FHL1-related diseases.9, 10, 11, 12, 13, 14, 15, 16, 17, 21 The available genotype–phenotype data indicate that patients belonging to the ‘RB subgroup’ harbour missense mutations or short in-frame deletions affecting the second LIM domain, whereas mutations found in XMPMA and EDMD/EDMD-like patients are either missense, in-frame or out-of-frame mutations localized in the more distal exons encoding the third and fourth LIM domains.17, 21 Histidine 123 and cysteine 224 appear as the main mutation hot spots for RBM and XMPMA, mutated in 7/15 and 5/9 families, respectively. The mutation found in the three British families results in the insertion of an isoleucine between Phe127 and Thr128, a two-amino-acid region (C–X2–C), linker between the two zinc-fingers of the second LIM domain and typically invariant in length. The insertion of a large aliphatic hydrophobic isoleucine between the rigid aromatic Phe127 and the hydrophilic Thr128 possibly disrupts the protein conformation by causing alteration of the orientation and spatial relationship between the tandem zinc-fingers.9, 22, 23
Although menadione–NBT staining was not performed in any of our patients and we cannot formally rule out the presence of RB, the clinical phenotype of our patients was not consistent with a diagnosis of RBM. In agreement with this finding, mutations affecting the LIM2 domain were reported also in three EDMD patients without RB,17 suggesting that the different type of mutations observed in these patients (small or large in-frame or out-of-frame deletions/insertions) might be responsible for the possible absence of RB in these patients.
Genotyping results showed segregation of an identical haplotype at the FHL1 locus in six affected individuals from the three families. Search of genealogic records did not permit us to trace the origin of the families to a common ancestral couple. However, haplotype analysis strongly suggested that the detected FHL1 mutation could represent a British founder mutation. In agreement with this finding, the p.Phe127_Thr128InsIle FHL1 mutation has never been reported in patients with a different ethnic background.
The observation of an identical gene mutation and haplotype in patients with different phenotypes indicates lack of genotype–phenotype correlation for this mutation and implies a possible role of additional factors in determining disease severity. Digenism has been demonstrated as the cause of diverging clinical phenotypes in association with the same mutation.24 However, in our case, this would implicate a second mutation event segregating with the FHL1 haplotype occurred in one of the descendents of the common ancestor. Immunohistochemical analysis of RBM patients’ muscle indicated that clinical severity might be correlated with the amount of residual FHL1 protein, and differential reduction in FHL1 amount was also observed in affected males and females from a SPM family with FHL1 mutation.10, 12 Unfortunately, the amount of FHL1 protein could not be assessed on muscle biopsies from our patients and further studies are warranted.
In conclusion, we identified three apparently unrelated UK families with a partly heterogenous clinical phenotype, in particular for disease onset, muscle involvement and disease progression, sharing a common FHL1 haplotype and mutation. Our findings suggest that the p.Phe127_Thr128insIle mutation in the FHL1 gene is a British founder mutation and FHL1 gene screening, in particular of exon 6, should therefore be indicated in British patients with a broad phenotypic spectrum of X-linked muscle diseases.
References
Freyd G, Kim SK, Horvitz HR : Novel cysteine-rich motif and homeodomain in the product of the Caenorhabditis elegans cell lineage gene lin-11. Nature 1990; 344: 876–879.
Dawid IB, Toyama R, Taira M : LIM domain proteins. C R Acad Sci III 1995; 318: 295–306.
Curtiss J, Heilig JS : DeLIMiting development. Bioessays 1998; 20: 58–69.
Jurata LW, Gill GN : Structure and function of LIM domains. Curr Top Microbiol Immunol 1998; 228: 75–113.
Kadrmas JL, Beckerle MC : The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 2004; 5: 920–931.
Brown S, McGrath MJ, Ooms LM, Gurung R, Maimone MM, Mitchell CA : Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear-cytoplasmic communication. J Biol Chem 1999; 274: 27083–27091.
Ng EK, Lee SM, Li HY et al: Characterization of tissue-specific LIM domain protein (FHL1C) which is an alternatively spliced isoform of a human LIM-only protein (FHL1). J Cell Biochem 2001; 82: 1–10.
McGrath MJ, Cottle DL, Nguyen MA et al: Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem 2006; 281: 7666–7683.
Windpassinger C, Schoser B, Straub V et al: An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet 2008; 82: 88–99.
Quinzii CM, Vu TH, Min KC et al: X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet 2008; 82: 208–213.
Chen DH, Raskind WH, Parson WW et al: A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci 2010; 296: 22–29.
Schessl J, Zou Y, McGrath MJ et al: Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest 2008; 118: 904–912.
Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I : Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology 2009; 72: 375–376.
Schessl J, Columbus A, Hu Y et al: Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics 2010; 41: 43–46.
Shalaby S, Hayashi YK, Goto K et al: Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1). Neuromuscul Disord 2008; 18: 959–961.
Gueneau L, Bertrand AT, Jais JP et al: Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet 2009; 85: 338–353.
Knoblauch H, Geier C, Adams S et al: Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol 2010; 67: 136–140.
Selcen D : Myofibrillar myopathies. Curr Opin Neurol 2010; 23: 477–481.
Anderson LV, Davison K : Multiplex western blotting system for the analysis of muscular dystrophy proteins. Am J Pathol 1999; 154: 1017–1022.
Klinge L, Aboumousa A, Eagle M et al: New aspects on patients affected by dysferlin deficient muscular dystrophy. J Neurol Neurosurg Psychiatry 2010; 81: 946–953.
Schoser B, Goebel HH, Janisch I et al: Consequences of mutations within the C terminus of the FHL1 gene. Neurology 2009; 73: 543–551.
Cowling BS, Cottle DL, Wilding BR, D’Arcy CE, Mitchell CA, McGrath MJ : Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromuscul Disord 2011; 21: 237–251.
Kloiber K, Weiskirchen R, Kräutler B, Bister K, Konrat R : Mutational analysis and NMR spectroscopy of quail cysteine and glycine-rich protein CRP2 reveal an intrinsic segmental flexibility of LIM domains. J Mol Biol 1999; 292: 893–908.
Muntoni F, Bonne G, Goldfarb LG et al: Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain 2006; 129: 1260–1268.
Acknowledgements
Diagnostic facilities at the Newcastle Muscle Centre are supported by the National Commissioning Group (NCG) for rare neuromuscular disorders. The Institute of Human Genetics in Newcastle is part of the MRC centre for Neuromuscular Diseases. VS and HL are members of the German Muscular Dystrophy Network (MD-NET 01GM0887) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany); http://www.md-net.org. Newcastle University and MD-NET are partner organisations of TREAT-NMD (EC, 6th FP, proposal #036825; http://www.treat-nmd.eu). We would like to thank the patients who took part in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sarkozy, A., Windpassinger, C., Hudson, J. et al. Phenotypic heterogeneity in British patients with a founder mutation in the FHL1 gene. Eur J Hum Genet 19, 1038–1044 (2011). https://doi.org/10.1038/ejhg.2011.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.84
Keywords
This article is cited by
-
A Dominant C150Y Mutation in FHL1 Induces Structural Alterations in LIM2 Domain Causing Protein Aggregation In Human and Drosophila Indirect Flight Muscles
Journal of Molecular Neuroscience (2021)
-
Diagnosis of muscle diseases presenting with early respiratory failure
Journal of Neurology (2015)
