
Abstract
Cone–rod dystrophies are inherited retinal dystrophies that are characterized by progressive degeneration of cones and rods, causing an early decrease in central visual acuity and colour vision defects, followed by loss of peripheral vision in adolescence or early adult life. Both genetic and clinical heterogeneity are well known. In a family with autosomal recessive cone–rod dystrophy, genetic analyses comprising genome scan with microsatellite markers, fine mapping and candidate gene approach resulted in the identification of a homozygous missense GUCY2D mutation. This is the first GUCY2D mutation associated with autosomal recessive cone–rod dystrophy rather than Leber's congenital amaurosis (LCA), a severe disease leading to childhood blindness. This study hence establishes GUCY2D, which is a common cause for both recessive LCA and dominant cone–rod dystrophy, as a good candidate for autosomal recessive cone–rod dystrophy.
Similar content being viewed by others


Introduction
Cone dystrophies (CODs) are a group of inherited pigmentary retinopathies that are characterized by degeneration of cones, causing an early decrease in visual acuity and colour vision in adolescence or early adult life. In many cases, the progressive nature of the clinical course reflects the peripheral vision loss, indicating rod degeneration. This may cause severe nyctalopia, and the condition is then called cone–rod dystrophy (CORD) (reviewed in Moore1and Hamel2). COD and CORD are genetically heterogeneous; several genes for dominant, recessive and X-linked forms have been identified by linkage and mutation analyses as listed in the University of Texas School of Public Health Retinal Information Network (http://www.sph.uth.tmc.edu/Retnet/), including ADAM9 (MIM 602713), AIPL1 (MIM 604392), CACNA1F (MIM 300110), CACNA2D4 (608171), CERKL3 (MIM 608381), CNGA34 (MIM 600053), CNGB34 (MIM 605080), CRX (MIM 602225), ABCA4 (MIM 601691), GUCA1A (MIM 600364), GUCY2D (MIM 600179), KCNV2 (MIM 607604), PDE6C (MIM 600827), PRPH2 (MIM 179605), RDH5 (MIM 601617), RIMS1 (MIM 606629), RPGRIP1(MIM 605446), SEMA4A (MIM 607292), PITPNM3 (MIM6 08921), PROM1 (MIM 604365) RAX2 (MIM 610362), RPGR (MIM 312610) and UNC119 (MIM 604011).
In this study, we have ascertained a consanguineous family with autosomal recessive CORD. Whole genome linkage analysis and fine mapping studies identified the sole homozygosity region on chromosome 17p13.3. GUCY2D, residing in this region, was selected as a strong candidate gene, because GUCY2D mutations have been reported as a frequent cause for both autosomal dominant CORD5, 6 (CORD6 (MIM 601777)) and Leber's congenital amaurosis7, 8 (LCA1 (MIM 204000)), which is typically a recessive condition. Indeed, we identified a novel recessive GUCY2D mutation in the family. This is the first recessive GUCY2D mutation associated with an autosomal recessive form of CORD rather than LCA.
Materials and methods
Subjects
A consanguineous Turkish family having six members afflicted with an autosomal recessive form of CORD was available for study (Figure 1). Blood samples for seven individuals were kindly provided by Dr Davut Gul at Gulhane Military Academy of Medicine in Ankara, and an additional seven samples plus repeated samples for affected individuals (patients 409, 507 and 509) were obtained later by the authors. DNA was extracted from whole blood using the salting out method with ammonium acetate. The study was approved by Boğaziçi University Committee on Research with Human Participants.

Partial pedigree diagram and haplotypes at 17p13.3–p11.2. Disease haplotype is boxed. Deduced alleles are in italics. DNA available for the genome scan is marked with a plus sign, and DNA available later is marked with an asterisk.
Detailed ophthalmologic examinations, including visual acuities, biomicroscopy, intraocular pressures and fundus examination after pupil dilation by mydriatics, were conducted on all patients. Fundus colour pictures were taken with Topcon Imagenet System. Electroretinography (ERG) and electrooculography (EOG) were performed using TOMEY EP 1000 Pro (Erlangen, Germany). ERG was performed and interpreted by the guidelines provided by the International Society for Clinical Electrophysiology of Vision (ISCEV). It was recorded after 20 min of full dark adaptation. Colour vision was tested by Ishihara Colour Blindness Test.
Molecular genetic studies
Genotyping and statistical analyses
A genome scan for 12 members of the family was performed at NHLBI (National Heart, Lung and Blood Institute) Mammalian Genotyping Service (Contract Number HV48141) using Marshfield Screen Set 16.9 The set contained 402 polymorphic microsatellite markers that spanned autosomes and sex chromosomes with an average density of 10 cM. The results of the genome scan were obtained in the LINKAGE file format and analyzed under the model of recessive inheritance, full penetrance and a disease gene frequency of one in 10 000. Software package easyLINKAGE10 was used for detecting and removing Mendelian inconsistencies (PedCheck),11 calculating two-point (SuperLink) and multipoint (SimWalk2) lod scores and constructing haplotypes (SimWalk2).12 The only suggestive locus 17p13.3 was further analyzed in our laboratory using 10 additional markers and also including two additional family members (Figure 1).
Mutational analyses
All coding exons and flanking sequences of GUCY2D (reference sequence: NM_000180.3) were analyzed for mutations, at least 20 nucleotides into introns. Intronic primers were designed using software Primer313 accessed through the Biology WorkBench website (http://workbench.sdsc.edu), and genome uniqueness was ascertained by in silico PCR (UCSC Genome Browser website, http://genome.ucsc.edu). Primer sequences and PCR conditions are available upon request. The amplified gene fragments were subjected to single-strand conformational polymorphism (SSCP) analysis and DNA sequencing as detailed elsewhere.14 Screening the family members and the population control group for the identified GUCY2D mutation c.2846T>C was performed by either SSCP analysis or high-resolution melting curve analysis in LightCycler 480 system (Roche Applied Science, Penzberg, Germany).
Results
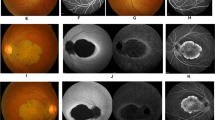
All six patients (31–70 years old) in the family were clinically investigated, and the disease was diagnosed as CORD according to clinical manifestations and electrophysiological findings that are summarized in Table 1. Dramatically reduced visual acuity, dyschromatopsia and nyctalopia observed in all affected individuals, together with variable presentation of photophobia, bone spicule formation, retinal vessel attenuation, retinal degeneration, macular and chorioretinal atrophy with decreased cone and rod responses during ERG, fulfilled the diagnostic criteria for CORD.2 EOG results were variable: in two patients they were below normal in both eyes and in the eldest patient above normal in both eyes. None of the patients was blind at birth, but they had reduced but stable vision loss according to family history. Patient 409 complained of progressive vision loss after the age of 60. Colour fundus and cone and rod ERG pictures of selected cases are presented in Figure 2.

(a) Colour fundus pictures of patient 509 showing the posterior pole (left eye) with macular atrophy and chorioretinal atrophy starting from midperipheral region of the retina (i), and the nasal region (left eye) with peripheral chorioretinal atrophy (seven-dot star), sparing midperipheral retina (four-dot star). The arrows disclose the sharp margin of normal-appearing and atrophic retina (ii). (b) Colour fundus pictures of patient 512 showing the posterior pole (right eye) with macular atrophy, retinal vessel attenuation and severe peripheral chorioretinal atrophy with bone spicule pigmentation, sparing midperipheral retina (iii), and the posterior pole (left eye) with macular atrophy, retinal vessel attenuation and severe peripheral chorioretinal atrophy with bone spicule pigmentation, sparing midperipheral retina (iv). (c) Cone ERG of patient 512 disclosing subnormal response (upper recording right eye and lower recording left eye) (v) and rod ERG of the same patient disclosing subnormal response (upper recording right eye, lower recording left eye; arrow a represents a-wave, and arrow b represents b-wave of rod ERG; vi).
Genetic findings
Genome-wide multipoint lod score analysis of the initial genotyping data assuming autosomal recessive inheritance and full penetrance yielded a single locus at chromosome 17p13.3 with a significant lod score of 4.23. Additional genotyping with 10 markers in all family members refined the gene locus to a 5.34-Mb interval between markers D17S1828 and D17S1791 (Figure 1), with maximum two-point and multipoint lod scores of 4.57 and 5.48, respectively (Table 2 and Figure 3). Crossover events in two regions, two recent ones around D17S559 in patients 509 and 511 and an ancestral one between D17S1844 and D17S1791, delineated the borders of the maximum homozygosity region. The haplotype data indicated identity by descent for the haplotype associated with the disease gene in the affected individuals. Among the 198 genes (NCBI Build 36.3) residing at this gene locus, GUCY2D was selected as a promising candidate and screened for mutations. All six affected individuals were found homozygous for a T>C transition in exon 15 (Figure 4), which segregated with the ocular phenotypes in the pedigree.

Multipoint lod scores of the total data set generated by the genome scan in a recessive model with full penetrance. Only lod scores >0 are plotted, in the order of chromosomal position. Top five scores are given within the graph. Multipoint lod score graph at the 17.21-Mb region on chromosome 17p13.3–p11.2 including markers used in fine mapping is enlarged in the lower panel.

Schematic representation of the c.2846T>C; p.Ile949Thr mutation together with sequence and segregation analyses. (a) A 1.2-Mb region at 17p13.1 including gene GUCY2D. Mutation c.2846T>C in exon 15 corresponds to a missense change (p.Ile949Thr) in the catalytic domain. Untranslated regions are indicated with open boxes. (b) Partial chromatograms showing c.2846T>C transition. (c) SSCP results for c.2846T>C screening in the family and four controls (C1–C4).
At the protein level, mutation c.2846T>C leads to the substitution of strongly hydrophobic isoleucine with polar threonine (p.Ile949Thr) in the catalytic domain (Figure 5a). Isoleucine at position 949 is highly conserved across species, with a rare replacement involving methionine in zebrafish (Figure 5b). This substitution was not detected in 186 control chromosomes screened, which corresponded to a power of 80% to detect a normal sequence variant with a frequency of 0.01.15 In addition, the substitution was predicted to be damaging by programs SIFT (sorting intolerant from tolerant, http://sift.jcvi.org/) and SNPs3D (http://www.snps3d.org/).

Structural context of p.Ile949Thr substitution and sequence conservation of Ile949. (a) Theoretical homodimer structure of retGC catalytic domain. The positions of mutated Ile949 residues in alpha helices together with bound Me2+ cofactors and GTP substrates are indicated with arrows, and their atomic structures are shown in blue. The structure was modelled using the Protein Workshop tool (Protein Data Bank, accession ID: 1AWL). (b) ClustalW multiple sequence alignments showing the evolutionary conservation of residue Ile949 (highlighted in grey) among human (Homo sapiens), dog (Canis familiaris), cattle (Bos taurus), mouse (Mus musculus) and rat (Rattus norvegicus), but in zebrafish (Danio rerio) a methionine residue is present at this position. Purple, neutral-polar; red, basic-polar; grey, neutral-nonpolar and blue, acidic-polar amino acids.
Discussion
Using whole genome linkage analysis followed by a positional candidate gene approach, we identified GUCY2D, encoding retina-specific guanylyl cyclase (retGC), as a causative recessive gene for autosomal recessive CORD. Recessive GUCY2D mutations have been previously associated with LCA, which is the most severe form of inherited retinopathies and a common cause of childhood blindness. This study links for the first time a recessive GUCY2D mutation with a new phenotype, CORD. In this gene, CORD has been associated with only dominant mutations so far.
The differential diagnosis of LCA and CORD is often not difficult. Total blindness or severely impaired visual function detected at birth or during early infancy, normal fundus and extinguished ERG are the key diagnostic findings for LCA (clinical manifestations reviewed in Allikmets16 and Hanein et al17). However, an early decrease in visual acuity with frequent dyschromatopsia, followed by progressive peripheral visual field deficits, which are called scotoma, are the main clinical entities of CORD. The six patients presented in this study, who had reduced but stable vision in adulthood, fulfil these diagnostic criteria for CORD (Table 1). Unlike LCA, they had subnormal but detectable ERG with the following abnormal fundus findings: pigment epithelial alterations were initially seen at the macular region and later noticed at the peripheral retina, sparing however the midperipheral retina, in which chorioretinal atrophy and bone spicule formation could be observed (Figure 2a and b).
In this study we identified a novel GUCY2D variant c.2846T>C; p.Ile949Thr that segregated with the ocular phenotype in the pedigree. We assessed that it was a mutation, as this change was absent in control chromosomes tested and the residue was evolutionarily conserved. In a theoretical model for the catalytic domain of retGC18 based on the crystal structure of the catalytic domain of type II adenylyl cyclase,19 949-isoleucine is positioned within an α-helical region on the outer surface. The substitution of isoleucine, which is the most hydrophobic residue, with polar threonine in this region would be expected to interfere with the proper folding of the helical segment, affecting the function of the catalytic domain. However, we propose that p.Ile949Thr does not abolish but only decreases the enzymatic activity, based on the extensive in vitro studies involving functional consequences of LCA missense mutations positioned in the catalytic domain of retGC.20, 21 Those missense mutations were all shown to be null alleles that dramatically impaired the catalytic activity and thus led to a severe phenotype, similar to the ones caused by truncating mutations that almost make up half of all GUCY2D mutations.7, 17
In all, 12% of all LCA types8 and up to 40% of dominant CORD6 are caused by defects in GUCY2D, an important component in the recovery process of phototransduction in the vertebrate retina. Almost all CORD6 mutations (all with dominant effect) are confined to three consecutive codons in exon 13 encoding part of the dimerization domain of retGC.6 Recessive GUCY2D mutations causing LCA1, in contrast, are distributed throughout the remaining exons.7 Mutation p.Ile949Thr identified in this study contrasts with the published results in the sense that (1) it resides in the catalytic domain but pathogenic in the homozygous state only, (2) it leads to recessive and not dominant CORD and (3) in homozygous state, it does not lead to LCA. Our finding establishes that recessive CORD can result from GUCY2D mutations, and we suggest screening of this gene for families afflicted with the recessive form of this group of disorders. This report is not only expected to benefit genetic diagnosis in families with CORD, but it also raises a caution against excluding known dominant CORD genes in families with recessive inheritance.
Accession codes
References
Moore A : Cone and cone-rod dystrophies. J Med Genet 1992; 29: 289–290.
Hamel C : Cone rod dystrophies. Orphanet J Rare Dis 2007; 2: 7.
Aleman T, Soumittra N, Cideciyan A et al: CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci 2009; 50: 5944–5954.
Thiadens A, Roosing S, Collin R et al: Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3 in progressive cone dystrophy. Ophthalmology 2010; 117: 825–830.e1.
Kelsell R, Gregory-Evans K, Payne A et al: Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum Mol Genet 1998; 7: 1179–1184.
Kitiratschky V, Wilke R, Renner A et al: Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest Ophthalmol Vis Sci 2008; 49: 5015–5023.
Perrault I, Rozet J, Gerber S et al: Spectrum of retGC1 mutations in Leber's congenital amaurosis. Eur J Hum Genet 2000; 8: 578–582.
den Hollander A, Roepman R, Koenekoop R, Cremers F : Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 2008; 27: 391–419.
Weber J, Broman K : Genotyping for human whole-genome scans: past, present, and future. Adv Genet 2001; 42: 77–96.
Lindner T, Hoffmann K : easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics 2005; 21: 405–407.
O’Connell J, Weeks D : PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63: 259–266.
Sobel E, Lange K : Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Corut A, Senyigit A, Ugur S et al: Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet 2006; 79: 650–656.
Collins J, Schwartz C : Detecting polymorphisms and mutations in candidate genes. Am J Hum Genet 2002; 71: 1251–1252.
Allikmets R : Leber congenital amaurosis: a genetic paradigm. Ophthalmic Genet 2004; 25: 67–79.
Hanein S, Perrault I, Gerber S et al: Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23: 306–317.
Liu Y, Ruoho A, Rao V, Hurley J : Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc Natl Acad Sci USA 1997; 94: 13414–13419.
Zhang G, Liu Y, Qin J et al: Characterization and crystallization of a minimal catalytic core domain from mammalian type II adenylyl cyclase. Protein Sci 1997; 6: 903–908.
Rozet J, Perrault I, Gerber S et al: Complete abolition of the retinal-specific guanylyl cyclase (retGC-1) catalytic ability consistently leads to Leber congenital amaurosis (LCA). Invest Ophthalmol Vis Sci 2001; 42: 1190–1192.
Tucker C, Ramamurthy V, Pina A et al: Functional analyses of mutant recessive GUCY2D alleles identified in Leber congenital amaurosis patients: protein domain comparisons and dominant negative effects. Mol Vis 2004; 10: 297–303.
Acknowledgements
We thank the family for participating in the study and Dr Davut Gul for the initial blood samples. We gratefully acknowledge the genome scan of the family performed by NHLBI Mammalian Genotyping Service (Contract Number HV48141). This work was supported by the Turkish State Planning Organization and Dünya Eye Hospital. AT was partially supported by the Turkish Academy of Sciences. SAU was a fellow of the Scientific and Technological Research Council of Turkey.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ugur Iseri, S., Durlu, Y. & Tolun, A. A novel recessive GUCY2D mutation causing cone–rod dystrophy and not Leber's congenital amaurosis. Eur J Hum Genet 18, 1121–1126 (2010). https://doi.org/10.1038/ejhg.2010.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.81
Keywords
This article is cited by
-
Variabilities in retinal function and structure in a canine model of cone-rod dystrophy associated with RPGRIP1 support multigenic etiology
Scientific Reports (2017)
-
GUCY2D mutations in a Chinese cohort with autosomal dominant cone or cone–rod dystrophies
Documenta Ophthalmologica (2015)
-
Genetic analysis of a consanguineous Pakistani family with Leber congenital amaurosis identifies a novel mutation in GUCY2D gene
Journal of Genetics (2014)
