
Abstract
PRKAG2 cardiac syndrome is an autosomal dominant inherited disease resulted from mutations in the PRKAG2 gene that encodes γ2 regulatory subunit of AMP-activated protein kinase. Affected patients usually develop ventricular tachyarrhythmia and experience progressive heart failure that is refractory to medical treatment and requires cardiac transplantation. In this study, we identify a H530R mutation in PRKAG2 from patients with familial Wolff-Parkinson-White syndrome. By generating H530R PRKAG2 transgenic and knock-in mice, we show that both models recapitulate human symptoms including cardiac hypertrophy and glycogen storage, confirming that the H530R mutation is causally related to PRKAG2 cardiac syndrome. We further combine adeno-associated virus-9 (AAV9) and the CRISPR/Cas9 gene-editing system to disrupt the mutant PRKAG2 allele encoding H530R while leaving the wild-type allele intact. A single systemic injection of AAV9-Cas9/sgRNA at postnatal day 4 or day 42 substantially restores the morphology and function of the heart in H530R PRKAG2 transgenic and knock-in mice. Together, our work suggests that in vivo CRISPR/Cas9 genome editing is an effective tool in the treatment of PRKAG2 cardiac syndrome and other dominant inherited cardiac diseases by selectively disrupting disease-causing mutations.
Similar content being viewed by others

Introduction
The Wolff-Parkinson-White (WPW) syndrome is a common cause of paroxysmal supraventricular tachycardia. With a prevalence of 0.1%-0.3%, WPW syndrome accounts for about 10.5% of sudden cardiac deaths in individuals younger than 35-years old1,2. WPW patients usually exhibit cardiomyopathy characterized by ventricular pre-excitation, progressive conduction system disease and cardiac hypertrophy3,4,5,6,7,8,9, followed later by fatal ventricular tachyarrhythmia and progressive heart failure refractory to medical treatment, leaving patients with cardiac transplantation as the only hope.
WPW syndrome can be caused by the mutations in the PRKAG2 gene that encodes the γ2 subunit of AMP-activated protein kinase (AMPK). As a heterotrimeric complex composed of a catalytic α-subunit and regulatory β- and γ-subunits, AMPK senses energy and nutrition in response to AMP/ATP fluctuations10. Energy deficiency elevates the cellular AMP/ATP ratio, which triggers AMP to bind the γ-subunit, thus inducing a conformational change and relieving α-subunit from autoinhibition. The exposed catalytic domain is then phosphorylated at Thr172 by upstream kinases, which activates AMPK and stimulates ATP-generating pathways while inhibits ATP-consuming pathways10,11. Mutations in PRKAG2, however, impair the sensitivity of AMPK to energy status, rendering glycogen-storage cardiomyopathy4,12,13. Intriguingly, suppressing the expression of transgenic PRKAG2 (N488I), a disease-causing mutation, during early postnatal stage successfully reverses the PRAKG2 syndrome14, suggesting an interesting possibility that the phenotypes of the PRKAG2-related WPW syndrome could be prevented if the mutant allele is disrupted while keeping the wild-type (WT) allele intact.
The prokaryotic type II clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome-editing system, coupling the Cas9 nuclease with short guide RNAs (sgRNAs)15,16,17,18, has been successfully applied to correct disease-causing mutations in the zygotes and livers19,20,21,22. In addition, adeno-associated virus (AAV)-based, CRISPR/Cas9-mediated excision of intervening DNA from the mutant dystrophin gene partially restored muscle structural and functional deficiencies in the mouse model of Duchenne muscular dystrophy (DMD)23,24,25. However, the potential possibility of using the CRISPR/Cas9 system to treat dominant inherited heart diseases such as PRKAG2 cardiac syndrome has yet to be evaluated.
In this study, we identified a novel H530R mutation in PRKAG2 from patients diagnosed with familial WPW syndrome. We demonstrate that the H530R mutation is causally related to PRKAG2 cardiac syndrome using heart-specific H530R-PRKAG2 transgenic and global knock-in mouse models. Strikingly, AAV9-mediated delivery of Cas9 nuclease coupled with sgRNAs targeting the mutant allele encoding H530R-PRKAG2 abolished the molecular defects in H530R-PRKAG2 transgenic and knock-in mice. A single systemic injection of AAV9-Cas9/sgRNA at postnatal day 4 (P4) or P42 effectively reversed cardiac hypertrophy and glycogen storage associated with PRKAG2 cardiac syndrome. In summary, our findings suggest that CRISPR/Cas9 genome editing may serve as a potential therapeutic approach to treat PRKAG2 cardiac syndrome and other dominant inherited cardiac diseases.
Results
Identification of H530R mutation in PRKAG2 in patients with familial WPW syndrome
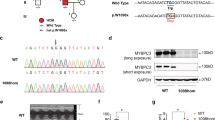
Clinical examinations revealed two subjects, a mother and a son, exhibiting conduction abnormalities and ventricular pre-excitation with a short PR interval (< 80 ms), a prolonged QRS duration (> 110 ms) and the presence of a slurred upstroke to the QRS complex (the delta wave) (Figure 1B and 1C). Echocardiography showed that the patients had left ventricular hypertrophy with ventricular wall thickness of > 20 mm (Figure 1A), consistent with increased voltages observed in electrocardiograms (Figure 1B). These manifestations were characteristics of PRKAG2-related familial WPW syndrome, such as ventricular pre-excitation, progressive conduction system disorder and cardiac hypertrophy. We next sequenced the PRKAG2 gene from the family and detected a heterozygous missense mutation c.1589A>G in the mother and son, but not the father who was diagnosed as normal (Figure 1C). This mutation resulted in an amino acid substitution of histidine with arginine at residue 530 (H530R) in PRKAG2 protein (Figure 1D). Further analysis of more members of this affected family (10 females and 9 males, aged 3 to 65 years at diagnosis) revealed that the grandfather also carried a heterozygous c.1589A>G mutation and exhibited conduction abnormalities and cardiac hypertrophy, whereas the others were healthy individuals with homozygous WT PRKAG2 gene (Figure 1C). These observations suggest that the H530R mutation is causally related to PRKAG2 cardiac syndrome and exhibits dominant inheritance. Interestingly, PRKAG2 H530R variant has been reported in one patient with a childhood-onset of cardiac hypertrophy26. However, the follow-up study on this case is lacking.

A missense mutation H530R of PRKAG2 is identified from a family with WPW syndrome. (A) Echocardiogram of the patient (the son). IVS, interventricular septum (16 mm); LVLW, lateral wall of left ventricle (22 mm); LA, left atrium; RA, right atrium; LV, left ventricle; RV, right ventricle. (B) 12-lead electrocardiogram of the patient (the son) with the PRKAG2 cardiac syndrome. (C) Pedigree of the family with PRKAG2 cardiac syndrome. Circles indicate female family members, and squares indicate male family members. Solid symbols denote affected family members, and the symbol with a slash indicates deceased family member. (D) Sequencing of PRKAG2 cDNA of the patient (the son). The arrow points to c.1589A>G mutation of PRKAG2 cDNA, which leads to missense mutation of H530R. (E) Diagram showing the heart-specific transgene constructs of PRKAG2. The copy numbers of exogenous PRKAG2 transgene were determined by qPCR analysis of αMHC promoter. (F) LV wall thickness calculated by echocardiography. Data represent mean ± SD. Two-way ANOVA was performed for overall differences (n = 5, 5, and 7 for control, Tg-WT, and Tg-H530R mice, respectively). Unpaired two-tailed Student's t-test was performed to analyze difference between Tg-H530R and Tg-WT mice (***P < 0.001). (G) Lead II electrocardiogram of 12-week-old mice. (H) Quantification of heart weight from 8-week-old control, Tg-WT, and Tg-H530R mice. Data represent mean ± SD. One-way ANOVA was used to analyze overall differences between all groups (n = 11, 8, and 18 for control, Tg-WT, and Tg-H530R mice, respectively). Unpaired two-tailed Student's t-test was performed for single comparison (***P < 0.001). (I) PAS staining of paraffin-embedded sections. (J) Measurement of glycogen content in hearts from 8-week-old control, Tg-WT and Tg-H530R mice. The values are 1.023 ± 0.203, 11.45 ± 0.865, and 63.06 ± 3.99 mg/g, respectively. Data represent mean ± SD. One-way ANOVA was performed for overall differences (n = 8, 5, and 12 for control, Tg-WT, and Tg-H530R mice, respectively). Unpaired two-tailed Student's t-test was performed for single comparison (***P < 0.001).
H530R mutation results in PRKAG2 cardiac syndrome
Transgenic mouse models have been widely used to validate the mutations causing PRKAG2 cardiac syndrome5,9,27,28. To investigate whether the H530R mutation accounts for the disease, we generated transgenic mice expressing WT (Tg-WT) and H530R (Tg-H530R) forms of PRKAG2 under the control of heart-specific α-myosin heavy chain (αMHC) promoter (Figure 1E).
Quantitative RT-PCR analysis revealed that WT and Tg-H530R mice carried 20 and 16 copies of the human PRKAG2 gene, respectively (Figure 1E). In addition, comparable level of exogenous human PRKAG2 protein was detected in the heart of both transgenic lines (Supplementary information, Figure S1A). Continuous echocardiography revealed that the left ventricular wall of Tg-H530R mice was significantly thicker than that of C57BL/6 (Control) or Tg-WT mice (Figure 1F). Tg-H530R mice, but not C57BL/6 or Tg-WT mice, had ventricular pre-excitation with a short PR interval and a wide QRS with the delta wave (Figure 1G). Moreover, a substantial increase in the heart weight was detected in Tg-H530R mice relative to C57BL/6 or Tg-WT mice at the age of 2 months (Figure 1H). Notably, we found the presence of large vacuoles in the cardiomyocytes of Tg-H530R mice (Supplementary information, Figure S1B). It was unlikely that these vacuoles were lipid droplets because the lipid content was remarkably reduced in Tg-H530R mouse heart (Supplementary information, Figure S1C-S1E). We further demonstrated that the cardiomyocytes were filled with PAS-positive material indicating glycogen (Figure 1I), and the myocardial glycogen content was dramatically increased in Tg-H530R mice (Figure 1J). In line with these findings, transmission electron microscopy also showed glycogen particles and vacuoles throughout the cardiomyocytes (Supplementary information, Figure S5D). In summary, we concluded that Tg-H530R mice recapitulated the clinical manifestations of patients with PRKAG2 cardiac syndrome.
We next generated a sequencing-validated knock-in heterozygous mouse line carrying only one nucleotide substitution resembling c.1589A>G mutation of human PRKAG2, thus causing a H527R mutation in mouse PRKAG2 equivalent to H530R in human PRKAG2 (Figure 2A-2C). It is referred to as H530R PRKAG2 knock-in (+/H530R) mouse model thereafter. We found that the left ventricular wall of +/H530R mice (0.768 ± 0.040 mm) was slightly thicker than that of WT controls (0.604 ± 0.026 mm) at 3 months of age (Figure 2D). Notably, the thickness of left ventricular wall of +/H530R mice was increased by ∼1.5-fold (0.921 ± 0.099 mm) compared with that in WT mice (0.629 ± 0.067 mm) by 6 months (Figure 2D). There was also a 40% increase in the heart size and weight of +/H530R mice (Figure 4A and 4B). Moreover, both PAS staining and enzymatic measurement showed significant glycogen accumulation in +/H530R mice (Figure 2E and 2F), similar to what was found in Tg-H530R mice. Together, evidence from transgenic and knock-in mice demonstrates that the H530R mutation of PRKAG2 was the cause of PRKAG2 cardiac syndrome. Interestingly, we obtained two independent lines that harbored the frame-shift mutation in one PRKAG2 allele (+/fs) during the generation of +/H530R mice (Supplementary information, Figure S3C-S3F). These mice (+/fs) appeared healthy, suggesting that one copy of WT PRKAG2 allele is sufficient for its normal function.

H530R knock-in mice recapitulate the PRAKG2 cardiac syndrome. (A) Schematic showing the generation of H530R-PRKAG2 knock-in mouse model using the CRISPR/Cas9 system. Lowercase characters denote the intronic region and uppercase characters denote the exonic region. (B) Alignment of genomic DNA and protein sequences of human and mouse PRKAG2 near the H530 locus. The H530 of human PRKAG2 is equivalent to H527 of mouse PRKAG2. The obtained knock-in mouse model is referred to as the H530R-PRKAG2 knock-in model. (C) Validation of heterozygous H530R knock-in mice by sequencing. (D) LV wall thickness calculated by continuous echocardiography. Data represent mean ± SD. Two-way ANOVA was performed for overall differences (n = 6). Unpaired two-tailed Student's t-test was performed for single comparison (***P < 0.001). (E) PAS staining of paraffin-embedded sections. (F) Measurement of glycogen content in hearts from 3-month-old WT (+/+) and heterozygous H530R knock-in (+/H530R) mice. Data represent mean ± SD. One-way ANOVA was performed for overall differences (n = 6). Unpaired two-tailed Student's t-test was performed for single comparison (***P < 0.001).

CRISPR/Cas9 system-mediated genome editing corrects PRKAG2 cardiac syndrome in the knock-in mouse model. (A) Heart images. (B) Heart weight statistics. (C) LV wall thickness measured by echocardiography. (D) PAS staining of paraffin-embedded sections. (E) Glycogen measurement. In B, C and E, one-way ANOVA was performed for overall differences between all groups (n = 6). Unpaired two-tailed Student's t-test was performed for single comparison (**P < 0.01, ***P < 0.001). (F) Transmission electron microscopy of ultrathin sections of hearts from 6-month-old mice as indicated.
Specificity of the designed CRISPR/Cas9 system
CRISPR/Cas9 is a powerful genome-editing tool16,17,18 and AAV9-mediated delivery preferentially targets cardiac cells29,30,31. We next attempted to combine these two approaches to target the PRKAG2 mutation associated with PRKAG2 cardiac syndrome. We first evaluated the efficacy and specificity of AAV9-mediated gene delivery following various administration routes. The EGFP-expressing AAV9 particles were injected into C57BL/6 mice and EGFP expression was analyzed by immunohistochemistry and immunoblotting 2 weeks post-injection. As shown in Supplementary information Figure S2A, we detected the lowest EGFP expression in the cardiac tissues following intrapericardial injection. Myocardial injection only yielded restricted EGFP expression. However, mice subjected to tail vein and intraventricular injections displayed much higher and more homogenous EGFP expression in the heart. A slightly higher EGFP expression was detected in the cohorts receiving intraventricular injection with aortic clamping. In addition, intraventricular injection of AAV9-EGFP at P4 or tail vein injection at P42 resulted in EGFP expression primarily in the heart (Supplementary information, Figure S2B-S2D). We therefore employed the systemic injection (tail vein or intraventricular) in the the following gene-editing studies.
We next cloned the Cas9 nuclease into a standard AAV9 vector32 and the sgRNAs into a self-complementary AAV9 (scAAV9) vector with fast and prolonged expression33. The ∼5.0 kb Cas9 expression cassette consisted of a CMV promoter, a 4.27-kb Cas9 coding sequence and a 48-bp miniature poly(A) signal, just reaching the upper-limit of the packaging capacity for AAV9 vector (Supplementary information, Figure S4A). Meanwhile, sgRNAs were designed by manually inspecting the genomic DNA sequence around the site encoding H530 for the NGG motif (Figure 3A). The human and mouse genomic DNA sequences around the site coding H530 are very similar (Figure 3A). Based on human PRKAG2 gene sequence, 4 sgRNAs targeting the mutant allele were selected for specificity test and 2 sgRNAs targeting the WT allele served as control. To determine the sgRNA specificity in human genome, we transfected the Cas9 nuclease and sgRNA candidates into WT human embryonic stem (ES) cells and the homozygous knock-in human ES cells with a single-nucleotide substitution (c.1589A>G, causing H530R mutation). Then the genomic DNA was analyzed by the T7E1 assay. We detected noticeable T7E1 digestion in the H530R mutant cells transfected with all 4 sgRNA candidates (Figure 3B). In WT ES cells, however, T7E1 digestion was observed with sgRNA-m1, sgRNA-m2 or sgRNA-m4 transfection but not with sgRNA-m3 transfection, suggesting the high specificity of sgRNA-m3. We further assessed the on-target and off-target indel frequencies of sgRNA-m3 in H530R knock-in ES cells using deep sequencing. The indel frequency was as high as 24% at the H530R site, whereas that at WT or other predicted off-target sites was barely detectable (0.4% and less, Figure 3C). Together, these results indicate that sgRNA-m3 specifically targets the mutant site of PRKAG2 in human genome encoding H530R.

CRISPR/Cas9 system specifically targets the H530R mutation site of PRKAG2 in human and mouse genome. (A) Schematic of candidate sgRNA target sites in human and mouse PRKAG2 gene. Lowercase characters denote the intronic region and uppercase characters denote the exonic region. PAM motifs (NGG) are highlighted in green. (B) Screening of sgRNAs by T7E1 assay in human WT and H530R knock-in ES cells. The selected sgRNA-m3 is highlighted in red. Blue arrows denote the digested DNA bands. (C) Frequency of indels by sgRNA-m3 at on-target (h-H530R) and off-target (h-OT1-h-OT10 and h-WT) sites in human WT and H530R ES cells. Data represent mean ± SD (n = 5). The most prevalent on-target indels of PRKAG2 H530R by sgRNA-m3 are shown in the lower panel. Line denotes the sgRNA sequence, and arrow denotes the Cas9 cleavage site. (D) Frequency of indels at on-target (m-H530R) and off-target (m-OT1-m-OT10 and m-WT) sites in heart genomic DNA from the +/H530R mouse injected with AAV9-Cas9/sgRNA-m3 at P4 or P42. Data represent mean ± SD (n = 6). Most prevalent on-target indels of PRKAG2 H530R in +/H530R mouse heart genomic DNA by sgRNA-m3 injected at P4 are shown in the lower panel. (E) qPCR analysis of PRKAG2 expression in the heart from mice as indicated. Data represent mean ± SD (n = 5). Unpaired two-tailed Student's t-test was performed for single comparison (**P < 0.01, ***P < 0.001). (F) Protein level analysis of AMPK subunits by immunoblots of heart tissues from mice as indicated.
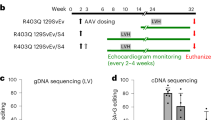
Since the sequence of sgRNA-m3-targeting site was exactly the same in human and mouse genome (Figure 3A), we next applied sgRNA-m3 directly to the animal studies. Premixed AAV9-Cas9 and scAAV9-sgRNA-m3 particles (AAV9-Cas9/sgRNA-m3) were delivered into +/H530R mice by intraventricular injection at P4 or intravenous injection at P42. Eight weeks after AAV injection, the heart was harvested and analyzed for indel frequencies at both on-target and off-target sites in the genome. Deep-sequencing results showed the indel of ∼6.5% and ∼2.6% at the H530R mutant locus in mice injected at P4 and P42, respectively. In addition, most indels were located at the Cas9-cleavage site and could produce frameshift mutations as revealed by the prevalent mutant sequences (Figure 3D). The indel frequency at the WT locus of PRKAG2 was 0.24%-0.26%, and at the other predicted off-target sites in mice injected at P4 and P42 was 0.24%-0.48% and 0.09%-0.32%, respectively (Figure 3D). These results indicate that sgRNA-m3 can recognize a single-nucleotide difference and selectively target the H530R mutation in mouse genome.
CRISPR/Cas9-mediated genome editing rescued PRKAG2 cardiac syndrome
We next investigated the effect of AAV9-Cas9/sgRNA-m3 treatment on cardiac PRKAG2 expression in +/H530R mice. About 20% reduction in the mRNA level was detected in mice receiving AAV9-Cas9/sgRNA-m3 injection on P4 or P42 (Figure 3E), suggesting that Cas9/sgRNA-m3 cleavage could cause nonsense-mediated mRNA decay. As a positive control, the mRNA level of +/fs mice was reduced to ∼65% of that of WT control. In addition, a modest increase in AMPKβ2 protein level and a dramatic decrease in AMPKα phosphorylation level were observed in the heart of +/H530R mice (Figure 3F), which were also observed in transgenic mice (Supplementary information, Figure S1A and S1F). These phenotypes resembled those of mice carrying R302Q mutation in PRKAG29,34, and were greatly reversed following Cas9/sgRNA-m3 injection. We also detected reduced AMPKγ2 protein level in +/H530R mice injected with Cas9/sgRNA-m3 relative to those with EGFP (Figure 3F, compare lanes 7-9 with lanes 4-6). Collectively, these results indicate that AAV9-Cas9/sgRNA-m3 can disrupt the mutant PRKAG2 allele and correct the molecular defect in +/H530R mice.
We next analyzed the therapeutic potential of CRISPR/Cas9-mediated genomic editing on PRKAG2 cardiac syndrome. +/H530R mice administered with AAV9-Cas9/sgRNA-m3 had a drastic reduction in the heart size and weight compared with those with AAV9-EGFP (Figure 4A and 4B). The thickness of the left ventricular wall was also significantly decreased in AAV9-Cas9/sgRNA-m3-injected mice (Figure 4C). In line with these findings, we observed a ∼70% decrease in myocardial glycogen content in +/H530R mice receiving AAV9-Cas9/sgRNA-m3 injection at P4 and P42 (Figure 4D and 4E). Moreover, AAV9-Cas9/sgRNA-m3-treated mice had regularly arranged myofibrils and much fewer vacuoles and glycogen particles as revealed by transmission electron microscopy (Figure 4F). In summary, the heart of +/H530R mice after receiving AAV9-Cas9/sgRNA-m3 resembled that of WT and +/fs mice (Figure 4B-4F).
We also generated a panel of sgRNAs targeting exogenous mutated PRKAG2 gene (Supplementary information, Figure S4B). Among those, sgRNA-1 exhibited the highest specificity for sequence encoding the H530R mutant but not WT PRKAG2 (Supplementary information, Figure S4C). We injected Tg-H530R mice intraventricularly with AAV9-Cas9/sgRNA-1 at P4 and examined the heart 8 weeks post-injection. Similar to our findings in +/H530R mice, symptoms of Tg-H530R mice, such as heart enlargement (Supplementary information, Figure S5A), glycogen accumulation (Supplementary information, Figure S5B-S5D), the presence of large vacuoles (Supplementary information, Figure S5B and S5D) as well as disorganized myofibrils (Supplementary information, Figure S5B and S5D), were all markedly reversed following AAV9-Cas9/sgRNA-1 injection. Moreover, typical signs of heart failure, such as progressively thickening of left ventricular wall and interventricular septum (Figure 5A and 5B), enlargement of left ventricular chamber (Figure 5C) and progressive reduction in the fractional shortening and ejection fraction of left ventricle (Figure 5D and 5E), were effectively rescued to the normal level in Tg-H530R mice treated with AAV9-Cas9/sgRNA-1. Finally, AAV9-Cas9/sgRNA-1 administration normalized the shortened PR interval and the broad QRS with the delta wave in adult Tg-H530R mice (Figure 5F) and reduced the incidence of ventricular pre-excitation by ∼40% (Figure 5G). These results strongly support that CRISPR/Cas9-mediated genomic editing can rescue PRKAG2 cardiac syndrome.

CRISPR/Cas9-mediated genome editing improves the heart function of PRKAG2 cardiac syndrome in transgenic mouse model. (A-E) Continuous measurement of LV wall thickness (A), the thickness of IVS (B), LV diastolic diameter (C), LV fractional shortening (D), and LV ejection fraction (E) by echocardiography. Two-way ANOVA was used to analyze overall differences between all groups (n = 31, 19, and 20 for control, AAV9-EGFP, and AAV9-Cas9/sgRNA-1-treated Tg-H530R mice, respectively, at age of 4 weeks; n = 28, 18, and 18 at age of 8 weeks; n = 18, 17, and 14 at age of 12 weeks; n = 18, 16, and 13 at age of 16 weeks; n = 14, 14, and 11 at age of 20 weeks; n = 12, 11, and 9 at age of 24 weeks, respectively). Unpaired two-tailed Student's t-test was performed for single comparison between AAV9-EGFP and AAV9-Cas9/sgRNA-1-treated Tg-H530R mice (*P < 0.05, **P < 0.01, ***P < 0.001). (F) Lead II electrocardiogram. (G) Incidence of pre-excitation of 12-week-old control, AAV9-EGFP and AAV9-Cas9/sgRNA-1-treated Tg-H530R mice.
Discussion
Like many other genetic cardiac diseases, PRKAG2 cardiac syndrome is an autosomal dominant disease resistant to traditional gene therapy that focuses on providing functional genes33,35. Dominant disorders mostly result from heterozygous gain-of-function mutations; and the treatment requires removal or disruption of the mutant allele. The CRISPR/Cas9 gene-editing system has been successfully used to correct mutations in mouse zygotes19,22 and to edit genes in the liver20,21. However, accurate repair of mutations has mostly been carried out in zygotes and embryos with a high efficiency of homologous recombination, which occurs infrequently in somatic tissues such as the cardiomyocytes; thus it has remained challenging to apply the CRISPR/Cas9 system to the heart. In the current study, we characterize a H530R mutation in PRKAG2 from familial WPW syndrome and demonstrate that this mutation is responsible for the disease by generating heart-specific H530R PRKAG2 transgenic and global knock-in mice. Furthermore, we successfully combined AAV9 and the CRISPR/Cas9 gene-editing system to disrupt the PRKAG2 mutant allele encoding H530R in the mouse heart. As a result, the symptoms PRKAG2 cardiac syndrome were dramatically improved in adult animals.
It has been reported that various mutations in the γ2 subunit of AMPK can cause PRKAG2 cardiac syndrome. Although these mutations result in similar phenotypes including glycogen storage, cardiac hypertrophy and ventricular pre-excitation, their influences on cardiac AMPK activity are distinct5,7,9,36. For example, the N488I and R531Q mutations constitutively increase AMPK activity5,7. R302Q first increases and then decreases the AMPK activity34. In R531G heart, the AMPK activity is normal in the first week and then decreases36. In our study, we observed a significantly lower AMPK activity in mice carrying the H530R mutation compared with WT. These observations indicate that the molecular linkage between AMPK activity and the syndrome is still obscure and requires further investigation. Although the genome-editing efficiency was relatively low (∼6.5% in mice injected at P4 and ∼2.6% in mice injected at P42), we observed significantly improved phenotypes in +/H530R mice receiving AAV9-Cas9/sgRNA-m3 injection (Figure 4A-4F). In Tg-H530R mice, the improvement was even more dramatic (Figure 5A-5G and Supplementary information, Figure S5A-S5D). Similar recent studies have also suggested that correction of a subset of cardiomyocytes can restore the function of the heart significantly.23,24,25. For instance, the phenotypes of mdx mice are significantly improved when dystrophin expression is rescued in 1.1 ± 1.1% and 3.2 ± 2.4% cardiomyocytes 4 and 8 weeks, respectively, following post-intraperitoneal injection of AAV-Cas9 sgRNAs23. In addition, low dystrophin levels (3%-15%) are sufficient to delay the onset of cardiomyopathy37. In our work, the genome-editing efficiency is comparable with or higher than that in the previous studies. These findings imply that a small genome-editing efficiency is sufficient for significant therapeutic effects.
CRISPR/Cas9-mediated exon-excision therapies partially recover functional dystrophin expression and rescue DMD in the muscle and the heart23,24,25. This approach works fine for the DMD mutations, however, not every gene remains functional after exon deletion. Also, exon-excision is not applicable to dominant mutations because it does not differentiate the WT allele from the mutant one. It is therefore critical to specifically disrupt only the dominant mutant allele while keeping the WT allele intact when using the CRISPR/Cas9 system to treat dominant disorders. Here, we selectively disable the dominant mutant allele while leaving the WT allele, which carries only one nucleotide difference, intact. Notably, we demonstrate that a single systemic injection at P4 or P42 result in drastic therapeutic effects in H530R PRKAG2 transgenic and knock-in mouse models. In summary, our study demonstrates that the CRISPR/Cas9 genome-editing system has great potential in treating various dominant genetic human diseases.
Materials and Methods
Reagents
Periodic acid-Schiff (PAS) staining system (#395B-1KT) was purchased from Sigma (St Louis, MO), Glycogen assay kit (#KA0861) from Abnova (Taipei), total cholesterol assay kit (20131112) and triglyceride assay kit (20131111) from Shanghai Kehua Bio-engineering Co, Ltd (Shanghai), phospholipids assay kit (PL7620) from Beijing Kinghawk Pharmaceutical Co, Ltd (Beijing), Surveyor Mutation Detection kit from Transgenomic (Omaha, NE), and Q5® High-Fidelity DNA polymerase and T7E1 from New England Biolabs (Ipswich, MA).
Antibodies
Primary antibodies used for immunoblotting were as follows: rabbit monoclonal antibodies against AMPKα (#2603S), AMPKβ2 (#4148S), AMPKγ2 (#2536S), and phospho-AMPKα (T172) (#2535S) were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibody against clathrin heavy chain (CHC) was from BD Transduction Laboratories (San Jose, CA). Mouse monoclonal antibody against Flag was from Sigma (St Louis, MO).
Animal study
Mice were fed on a chow diet ad libitum and housed in a pathogen-free animal facility in plastic cages at 22 °C, with a daylight cycle from 6 am to 6 pm. All animal procedures were approved by the Animal Care and Use Committee, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences.
Transgenic and knock-in mice
For transgenic mice, PRKAG2 cDNA was cloned into pcDNA3.0 from human cDNA using primer pair: PRKAG2-F: 5′-ATCGGATCCCTGGTTAGAGTTATGGGAAGCG3′, PRKAG2-R: 5′-ACGCTCGAGCGTCTACATTCACGGCGGTC-3′. His530Arg mutation was obtained by site-directed mutagenesis using primer pair: H530R-F, 5′-AAGAGCTGAGGTCCGTCGGCTGGTGGTGGTAAATG-3′; H530R-R, 5′-CACCAGCCGACGGACCTCAGCTCTTACTATTCT-3′. Transgenic plasmids were constructed by inserting the cDNA of WT and H530R mutant human PRKAG2 into SalI site of the vector pJG/ALPHA MHC38, which consisted of an αMHC promoter, multi-cloning site and 3′ human growth hormone polyA site. Transgene constructs that contained the α-MHC promoter, PRKAG2 cDNA, and 3′ UTR human growth hormone polyA were linearized using BamHI and injected into fertilized C57BL/6 mouse oocytes by Shanghai Research Center for Model Organisms (Shanghai, China). Transgenic mice were PCR identified using primer pairs: TG-1F, 5′-CAAGTGACCAAGCAGCACAC-3′; TG-1R, 5′-ACCGTTGGCTACCAAAGCAA-3′; TG-2F, 5′-ATATCTCCAGATGCAAGCCTCTTC-3′; TG-2R, 5′-GTCCACGATGGTCTCCAGTAT-3′. Mice with WT and mutant transgene were labeled as Tg-WT and Tg-H530R, respectively.
Heterozygous knock-in mice carrying the c.1580A>G mutation (His527Arg) of murine PRKAG2 were also generated by Shanghai Research Center for Model Organisms. Briefly, 5′-CTCTTTGCAGGTCCATCGGC-3′ was chosen as targeting guide RNA. mRNA of in vitro transcribed Cas9 and chimeric sgRNA as well as a 121-bp single-stranded oligodeoxynucleotide (ssODN) were injected into zygotes of C57BL/6 mouse, as illustrated in Figure 2A. Obtained F0 mice were validated by sequencing using primer pairs: mPRKAG2-F, 5′-GCCAGGGAGGCTAGTGTAGT-3′; mPRKAG2-R, 5′-CAAGCCGGGTTCGATTATGT-3′. Mice with expected single-nucleotide mutation was crossed with WT (+/+) C57BL/6 mice to produce F1 knock-in mice. And 2 strains with frameshift mutations (+1 and −7 nucleotides) were also obtained and crossed with WT C57BL/6 mice. The obtained knock-in mice resembling human PRKAG2 H530R mutation were referred to as H530R knock-in mice. Offspring with point mutation (H530R) and frameshift (fs) mutations were labeled as +/H530R and +/fs, respectively. Sequence of the ssOND for knock-in mice generation was: 5′-CAAATGCACCCAACACACAAACAGTAAACTCACACCTGGACTTTTTCTCTTTGCAGGTCCGTCGGCTGGTTGTAGTGAATGAAGCAGATAGCATTGTGGGTATTATCTCCCTGTCAGACAT-3′.
Generation of human knock-in H530R mutant cell line
Human H9 ES cells were used to generate knock-in cells carrying the c.1589A>G mutation of PRKAG2 gene with CRISPR/Cas9 system. gRNA was designed online and the highest ranking candidate was cloned into a modified pX330 vector (Dr Feng Zhang's lab, http://www.addgene.org/42230/) with mCherry expression cassette as previously described17,39. The plasmids and a 121-bp ssODN were transfected into human ES cells with Lipofectamine2000 and mCherry positive cells were sorted by FACS 24 h later. Cells were then seeded into 96-well plates at a concentration of < 0.5 cell/well. Clones derived from single cell were validated by sequencing using primer pair hPRKAG2-F, 5′-GTAGAGATGATGTCTCACTGC-3′; hPRKAG2-R, 5′-TGATGGTTTAAATGCTGCACTTC-3′. The pure knock-in mutant clone was cultured for subsequent analysis. The sequence of the 121-bp ssODN for human knock-in cell preparation was: 5′-CATTTTAAACCTTTTTTAAATGTTAATTTTCACGTGTCCTGCTTTTCTCTTTGCAGGTCCGTCGGCTGGTGGTGGTAAATGAAGCAGATAGTATTGTGGGTATTATTTCCCTGTCGGACAT-3′.
Glycogen assay
Heart tissue was snap-frozen in liquid nitrogen. Glycogen was extracted and measured according to user's manual (Abnova, KA0861).
Histochemistry
Mouse heart tissue was fixed in 4% PFA and embedded in paraffin. Subsequently, 3 μm paraffin sections were prepared. Samples were deparaffinized and stained with hematoxylin-eosin. For glycogen staining, deparaffinized sections were stained with PAS reagent and hematoxylin according to the user manual (Sigma, 395B-1KT).
Transmission electron microscopy
Mouse heart ventricular tissue was immediately fixed in 2.5% glutaraldehyde solution. Ultrathin sections of 70 nm were prepared and stained with uranyl acetate and lead citrate before transmission electron microscopy analysis.
Western blot
Heart tissue was snap-frozen before being homogenized in RIPA buffer containing protease and phosphatase inhibitors with a high-throughput tissue homogenizer (Bertin Technologies, Precellys24). The homogenate was centrifuged at 12 000 rpm for 10 min at 4 °C, and the supernatant was mixed with loading buffer after determination of the protein concentration according to Lowry method (Bio-Rad). The sample was boiled at 95 °C for 10 min, separated by SDS-PAGE, transferred to nitrocellulose membrane and subjected to immunoblot analysis with Tanon 5200 chemiluminescent imaging system (Shanghai).
Echocardiography
Transthoracic echocardiography, using iE33 (Philip), was performed on patients in this study. 2D and Doppler echocardiographic measurements, including left ventricular maximal wall thickness, left atrial and left ventricular dimensions at end-systole and end-diastole were performed in accordance with previously published methods40. The left ventricular ejection fraction was calculated according to Simpson's method41. When appropriate, peak resting and/or exercise-induced left ventricular outflow tract obstruction, were estimated by continuous-wave Doppler at rest and/or during exercise (bicycle).
For mice, echocardiographic analysis was performed by technician blinded to the experiments. Mice were isofurane anesthetized and analyzed using an animal specific instrument (VisualSonics Vevo770®; Visual Sonics Inc, Toronto, Canada) as previously described42. LV end-diastolic and end-systolic diameters and wall thickness were obtained from M-mode tracings from measurements averaged from 6 separate cycles. LV fractional shortening (in percentage) was derived by the equation fractional shortening = ((LV end-diastolic diameter − LV end-systolic diameter)/LV end-diastolic diameter) × 100.
Electrocardiogram
Surface 12-lead electrocardiogram was performed while patients were at rest at a paper speed of 25 mm/s. Electrocardiograms were analyzed for basic rhythm, QRS axis, PR interval, Q waves, QRS and QT intervals, and ST-segment changes.
For mice, 6-lead surface electrocardiograms were obtained from 8- to 12-week old mice anesthetized with 2% intraperitoneal pentobarbital (2.5 μl/g). ECG intervals including PR, QRS, and QT were measured for each mouse. Ventricular pre-excitation was diagnosed on the basis of a short PR interval (< 15 ms) with prolonged QRS duration (> 17 ms) as previously described14.
Screening of guide RNAs
Candidate guide RNAs were designed by visual inspection of the sequences based on the requirements of NGG motif and the guide RNA spanning the mutated H530R site. Candidate sgRNA were chosen and cloned into modified pX330 as previously described. The obtained plasmids were transfected into WT and H530R knock-in human ES cells. Forty-eight hours later, the cells with mCherry signal were sorted by FACS. DNA then was extracted and PCR amplified by Q5 high-fidelity DNA polymerase for T7E1 assays. The primers were: hPRKAG2-F, 5′-GTAGAGATGATGTCTCACTGC-3′; hPRKAG2-R, 5′-TGATGGTTTAAATGCTGCACTTC-3′. The gRNA that preferentially targeted only the mutant allele was preferred.
On-target and off-target mutagenesis analysis
The On-target and off-target effects of sgRNA candidates were analyzed by T7E1 assay in WT and H530R knock-in human ES cell lines described above. The sgRNA-m3 was selected for ongoing experiments. Off-target sites for selected sgRNA-m3 were predicted using the Optimized CRISPR design server (http://crispr.mit.edu/)39. The WT allele with one-base mismatch and 10 top-scoring sites were selected (Supplementary information, Table S1). Briefly, PCR with indexed primers was used to amplify the genomic regions flanking the predicted on-target and off-target sites from human and mouse genomic DNA. Purified DNA samples were quantified by Qubit 2.0 Fluorometer (Life Technologies) and pooled in an equimolar ratio. The obtained libraries were sequenced with 250 bp paired-end reads on an Illumina Miseq system (Wuxi Apptec, Shanghai). The sequencing reads were mapped using BWA (http://bio-bwa.sourceforge.net/) and insertion or deletion (indels) were called by samtools with custom scripts. For each on-target and off-target site, indels were counted in a 50-bp window centered on the Cas9 potential cleavage sites.
AAV9-mediated gene expression
The AAV cloning vector pAOV-CMV-mCherry was modified as illustrated in Supplementary information Figure S3A. Briefly, a modified vector pAOV-CMV-minipolyA was obtained by replacing the DNA fragment between CMV and right ITR with PCR amplified minipolyA and the flanking cloning sites. 3× Flag-Cas9 sequence was subcloned from pX330 into pAOV-CMV-minipolyA. For sgRNA expression, the D sequence of left ITR was deleted and the sequence between both ITR was replaced with the U6-chimeric guide RNA scaffold. sgRNA-1 or sgRNA-m3 was inserted into the modified vector. Constructs for both Cas9 and sgRNA were sent to Obio Technology (Shanghai) for AAV9 package and purification.
P4 mice were intraventricularly injected with AAV 9 using a 29-gauge insulin syringe (BD, Ultral fine needle). Each P4 mouse received a total volume of 50 μl containing 2.5 × 1011 vg AAV9-EGFP or 5 × 1011 vg AAV9-Cas9 and AAV9-sgRNA viruses. 6-week old +/h530R knock-in mice were intravenously injected with 100 μl containing 1 × 1012 vg AAV9-EGFP or 200 μl containing 3 × 1012 vg AAV9-Cas9 and 1 × 1012 vg AAV9-sgRNA-m3 viruses. 8 weeks after AAV injection, heart tissue from some mice was collected for DNA, RNA, protein, and glycogen analysis. At the age of 6 months, heart tissue from other groups was collected for weight, glycogen, and transmission electron microscopy analysis.
Statistics
Values are given as mean standard error. Unpaired two-tailed Student's t-tests were performed in Excel (Microsoft) to analyze single comparison between two groups. P < 0.05 was regarded as significant, and *P < 0.05, **P < 0.01, ***P < 0.001. ANOVA was used to analyze overall differences between all groups in Prism5 (GraphPad software).
Author Contributions
CX, BLS and YY conceived and designed the experiments. CX performed most of the experiments and analyzed data with the assistance from JH, DL, ZY, JZ and JX. YPZ and YY collected patient samples and analyzed data. LS and NJ constructed human H530R knock-in ES cell line. JLD helped with transmission electron microscopic analysis. CX, JL, WQ, and BLS wrote the manuscript with input from all other authors. All authors approved the final version of the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
Basso C, Corrado D, Rossi L, Thiene G . Ventricular preexcitation in children and young adults: atrial myocarditis as a possible trigger of sudden death. Circulation 2001; 103:269–275.
Gollob MH, Green MS, Tang AS, Roberts R . PRKAG2 cardiac syndrome: familial ventricular preexcitation, conduction system disease, and cardiac hypertrophy. Curr Opin Cardiol 2002; 17:229–234.
Gollob MH, Green MS, Tang AS, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 2001; 344:1823–1831.
Arad M, Benson DW, Perez-Atayde AR, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 2002; 109:357–362.
Arad M, Moskowitz IP, Patel VV, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 2003; 107:2850–2856.
Blair E, Redwood C, Ashrafian H, et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001; 10:1215–1220.
Burwinkel B, Scott JW, Buhrer C, et al. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet 2005; 76:1034–1049.
Arad M, Maron BJ, Gorham JM, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 2005; 352:362–372.
Sidhu JS, Rajawat YS, Rami TG, et al. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation 2005; 111:21–29.
Hardie DG, Ross FA, Hawley SA . AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251–262.
Zaha VG, Young LH . AMP-activated protein kinase regulation and biological actions in the heart. Circ Res 2012; 111:800–814.
Luptak I, Shen M, He H, et al. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest 2007; 117:1432–1439.
Zou L, Shen M, Arad M, et al. N488I mutation of the gamma2-subunit results in bidirectional changes in AMP-activated protein kinase activity. Circ Res 2005; 97:323–328.
Wolf CM, Arad M, Ahmad F, et al. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation 2008; 117:144–154.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R . One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013; 154:1370–1379.
Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013; 153:910–918.
Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339:819–823.
Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science 2013; 339:823–826.
Wu Y, Liang D, Wang Y, et al. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013; 13 :659–662.
Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 2014; 32:551–553.
Yang Y, Wang L, Bell P, et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol 2016; 34:334–338.
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN . Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014; 345:1184–1188.
Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016; 351:400–403.
Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016; 351:403–407.
Tabebordbar M, Zhu K, Cheng JK, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016; 351:407–411.
Morita H, Rehm HL, Menesses A, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 2008; 358:1899–1908.
Banerjee SK, McGaffin KR, Huang XN, Ahmad F . Activation of cardiac hypertrophic signaling pathways in a transgenic mouse with the human PRKAG2 Thr400Asn mutation. Biochim Biophys Acta 2010; 1802:284–291.
Ahmad F, Arad M, Musi N, et al. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation 2005; 112:3140–3148.
Inagaki K, Fuess S, Storm TA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 2006; 14:45–53.
Bostick B, Ghosh A, Yue Y, Long C, Duan D . Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther 2007; 14:1605–1609.
Bish LT, Morine K, Sleeper MM, et al. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum. Gene Ther 2008; 19:1359–1368.
Senis E, Fatouros C, Grosse S, et al. CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol J 2014; 9:1402–1412.
Daya S, Berns KI . Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev 2008; 21:583–593.
Folmes KD, Chan AY, Koonen DP, et al. Distinct early signaling events resulting from the expression of the PRKAG2 R302Q mutant of AMPK contribute to increased myocardial glycogen. Circ Cardiovasc Genet 2009; 2:457–466.
Cirino AL, Ho CY . Genetic testing for inherited heart disease. Circulation 2013; 128:e4–e8.
Davies JK, Wells DJ, Liu K, et al. Characterization of the role of gamma2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am J Physiol Heart Circ Physiol 2006; 290:H1942–H1951.
van PM, van der Pijl EM, Hulsker M, et al. Low dystrophin levels in heart can delay heart failure in mdx mice. J Mol Cell Cardiol 2014; 69:17–23.
Gulick J, Subramaniam A, Neumann J, Robbins J . Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem 1991; 266:9180–9185.
Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 2013; 31:827–832.
Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005; 18:1440–1463.
Stafford WJ, Trohman RG, Bilsker M, Zaman L, Castellanos A, Myerburg RJ . Cardiac arrest in an adolescent with atrial fibrillation and hypertrophic cardiomyopathy. J Am Coll Cardiol 1986; 7:701–704.
Sano M, Minamino T, Toko H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007; 446:444–448.
Acknowledgements
We thank Jian-Guo Jia (Zhongshan Hospital), Jie Xu, Jie Qin, Yu-Xiu Qu and Hong-Hua Miao (Shanghai Institute of Biochemistry and Cell Biology), and Yu Kong (Electron Microscopy Facility at the Institute of Neuroscience) for technical assistance. We thank Dr Jeffrey Robbins (Cincinnati Children's Hospital Medical Center) for the heart-specific transgenic plasmid. This work was supported by the Ministry of Science and Technology of China (2016YFA0500100 and 2014DFG32410), China Postdoctoral Science Foundation (2012M520951 and 2013T60474), the National Natural Science Foundation of China (31430044, 31230020 and 81270155), Shanghai Science and Technology Committee (15140901902), the Opening Project of Shanghai Key Laboratory of Complex Prescription (Shanghai University of Traditional Chinese Medicine) (14DZ2271000) and the Opening Project of State Key Laboratory of Neuroscience (Institute of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences; SKLN-201503). CX gratefully acknowledges the support of SA-SIBS Scholarship Program.
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
PRKAG2-H530R transgenic mice develop cardiac hypertrophy. (PDF 8874 kb)
Supplementary information, Figure S2
AAV9-mediated EGFP expression in C57BL/6 mice. (PDF 7840 kb)
Supplementary information, Figure S3
Sequencing of H530R knock-in mouse and frame-shift mutant mouse strains. (PDF 1687 kb)
Supplementary information, Figure S4
AAV9-delivered CRISPR/Cas9 system to neonatal transgenic hearts achieves mutagenesis at target mutation site. (PDF 520 kb)
Supplementary information, Figure S5
CRISPR/Cas9 system rescues the manifestations of PRKAG2 cardiac syndrome in transgenic mouse model. (PDF 7091 kb)
Supplementary information, Table S1
Predicted off-target sites of sgRNA-m3 in human and mouse genomes. (PDF 157 kb)
Supplementary information, Table S2
PCR Primers of predicted on-target and off-target sites of sgRNA-m3 for deep sequencing. (PDF 136 kb)
Supplementary information, Table S3
Predicted off-target sites of sgRNA-1 in mouse genome. (PDF 193 kb)
Supplementary information, Table S4
PCR Primers of predicted on-target and off-target sites of sgRNA-1 for surveyor assay. (PDF 135 kb)
Rights and permissions
About this article

Cite this article
Xie, C., Zhang, YP., Song, L. et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res 26, 1099–1111 (2016). https://doi.org/10.1038/cr.2016.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.101
Keywords
This article is cited by
-
The role of cardiac microenvironment in cardiovascular diseases: implications for therapy
Human Cell (2024)
-
Homology-directed repair of an MYBPC3 gene mutation in a rat model of hypertrophic cardiomyopathy
Gene Therapy (2023)
-
CRISPR/Cas-mediated genome editing in mice for the development of drug delivery mechanism
Molecular Biology Reports (2023)
-
Nogo-B promotes angiogenesis and improves cardiac repair after myocardial infarction via activating Notch1 signaling
Cell Death & Disease (2022)
-
In vivo genome editing in mouse restores dystrophin expression in Duchenne muscular dystrophy patient muscle fibers
Genome Medicine (2021)
