
Abstract
The SCF (SKP1 (S-phase-kinase-associated protein 1), Cullin-1, F-box protein) E3 ubiquitin ligases, the founding member of Cullin-RING ligases (CRLs), are the largest family of E3 ubiquitin ligases in mammals. Each individual SCF E3 ligase consists of one adaptor protein SKP1, one scaffold protein cullin-1 (the first family member of the eight cullins), one F-box protein out of 69 family members, and one out of two RING (Really Interesting New Gene) family proteins RBX1/ROC1 or RBX2/ROC2/SAG/RNF7. Various combinations of these four components construct a large number of SCF E3s that promote the degradation of many key regulatory proteins in cell-context, temporally, and spatially dependent manners, thus controlling precisely numerous important cellular processes, including cell cycle progression, apoptosis, gene transcription, signal transduction, DNA replication, maintenance of genome integrity, and tumorigenesis. To understand how the SCF E3 ligases regulate these cellular processes and embryonic development under in vivo physiological conditions, a number of mouse models with transgenic (Tg) expression or targeted deletion of components of SCF have been established and characterized. In this review, we will provide a brief introduction to the ubiquitin-proteasome system (UPS) and the SCF E3 ubiquitin ligases, followed by a comprehensive overview on the existing Tg and knockout (KO) mouse models of the SCF E3s, and discuss the role of each component in mouse embryogenesis, cell proliferation, apoptosis, carcinogenesis, as well as other pathogenic processes associated with human diseases. We will end with a brief discussion on the future directions of this research area and the potential applications of the knowledge gained to more effective therapeutic interventions of human diseases.
Similar content being viewed by others

Introduction
The ubiquitin-proteasome system (UPS) fulfills essential cellular functions in eukaryotes through timely degradation of a variety of regulatory proteins1. The UPS-targeted protein degradation involves two well-defined steps: (1) the covalent attachment of multiple ubiquitin molecules to a substrate and (2) the recognition and degradation of ubiquitin-tagged substrate by the 26S proteasome2,3. Covalent conjugation of ubiquitin to the substrate, a process called ubiquitylation, is carried out by a three-step enzymatic cascade, involving the E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase4 (Figure 1). The substrate specificity of ubiquitylation is determined by the E3 enzymes that directly bind to the target substrates5. E3 ubiquitin ligases can be further divided into four major classes: HECT (Homologous to E6-AP C-terminus) domain-containing E3s, RING (Really Interesting New Gene) finger-containing E3s, N-end rule E3s, and APC/C (Anaphase-Promoting Complex/Cyclosome) E3s4,6. Whereas the HECT catalytic domain forms an E3-ubiquitin thioester intermediate prior to transferring the ubiquitin to the bound substrate4,7, the RING E3s use a RING finger domain to recruit ubiquitin-loaded E2 and transfer the ubiquitin from the E2 to a lysine residue on a targeted substrate via an isopeptide bond8. Ubiquitin has seven lysine residues (K6, K11, K27, K29, K33, K48, and K63), and can be further conjugated by another ubiquitin to form polyubiquitin chains with distinct linkages. As shown in Figure 1, the tagged substrates destined for proteasome-mediated degradation usually have a K48-linked polyubiquitin chain, which are recognized by 26S proteasomes for targeted degradation4,9, whereas K63-linked polyubiquitin chains have been strongly implicated in regulating various proteasome-independent cellular functions, including NF-κB signaling10, ribosomal function11, DNA replication and repair12, and intracellular trafficking13. Furthermore, a substrate protein can also be conjugated with just a single ubiquitin, a process known as monoubiquitylation, which modulates various biological functions14.

The ubiquitin-proteasome system. Ubiquitin (Ub) is first activated in an ATP-dependent reaction by the E1 ubiquitin-activating enzyme, and transferred to the E2 ubiquitin-conjugating enzyme, and finally transferred to the substrate with the help of an E3 ubiquitin ligase, which binds both the substrate protein and the ubiquitin-charged E2 enzyme, resulting in the mono-, or polyubiquitylation of the substrate. The mode of ubiquitylation and ubiquitin linkage determine the fate of the substrate: either being modified with altered function (mono-ubiquitylation or K63-linked polyubiquitylation) or being degraded (for K48 linkage) by the 26S proteasome, which consists of catalytic 20S and regulatory 19S subunits.
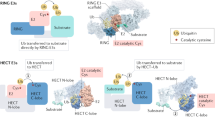
The Cullin-RING ligase (CRL) is the largest family of E3 ubiquitin ligases with eight family members, designated as CRL-1, CRL-2, CRL-3, CRL-4A, CRL-4B, CRL-5, CRL-7, and the much less known CRL-9, also designated as PARC (p53-associated parkin-like cytoplasmic protein)6,15,16,17. Each family member of CRL E3s consists of a scaffold protein cullin (eight family members), an adaptor protein (many family members), a RING protein (two family members), and a substrate receptor protein (many family members) (for review, see6) (Figure 2). The SCF E3 ubiquitin ligase, also designated as CRL-1, is the founding member of CRL E3. The SCF, consisting of SKP1 (S-phase-kinase-associated protein 1), Cullin-1/Cdc53, and an F-Box protein, was first defined by two independent groups in 1996-199718,19,20. Subsequent work published in 1999 from several laboratories revealed that a fully active SCF E3 requires a RING protein, RBX1/ROC1 (RING box protein 1/regulator of cullins 1)21,22,23,24 or its family member, RBX2/ROC2/SAG (Sensitive to Apoptosis Gene)25,26. Within the SCF E3 ligases, the cullin subunit cullin-1 (Cul-1) functions as a molecular scaffold that simultaneously interacts at its N-terminus with the SKP1-F-box complex, and at its C-terminus with RBX1 or RBX2, which binds to the ubiquitin-loaded E2 protein27 (Figure 2A). The F-box proteins, by binding to SKP1 and cullin-1 through the F-box domain, and to substrates through their WD40 or leucine-rich repeats (LRRs) domains, determine the substrate specificity of the SCF complex27,28. The cullin-1-RBX1 or cullin-1-RBX2 complex, with RBX1/2 binding to ubiquitin-loaded E2, constitutes the core E3 ligase activity29. By promoting timely ubiquitylation and subsequent degradation of more than 350 substrates30, the SCF E3 ligases have vital roles in the maintenance of the integrity of basic cellular processes, including signal transduction, gene transcription, DNA replication and repair, development, cell cycle progression, and preservation of genome stability31,32.

Schematic illustration of CRLs. (A) CRL-1, also known as SCF, is assembled together by a scaffold protein cullin-1, which binds at the N-terminus the adaptor protein SKP1 and an F-box protein, and at the C-terminus a RING protein RBX1 or RBX2/SAG that interacts with the ubiquitin-loaded E2. Generally, most targeted substrates have to be phosphorylated by a kinase(s) before being recognized by an F-box protein. SCF E3, whose activity requires cullin-1 neddylation, catalyzes the ubiquitin transfer from the E2 to the phosphorylated substrate. (B) CRL-2 consists of cullin-2, RBX1, adaptor proteins Elongin B/C and the substrate receptor VHL. (C) CRL-3 consists of cullin-3, RBX1, and the substrate receptor BTB (Bric-a-brac, Tram-track, Broad-complex domain). (D) CRL-4A/B consists of the cullin-4A or cullin-4B, RBX1, adaptor protein DDB1 (DNA damage-binding protein 1), and the substrate receptor DCAF (DDB1-CUL4-associated factor). (E) CRL-5 consists of cullin-5, RBX2, adaptor proteins Elongin B/C, and the substrate receptor SOCS. (F) CRL-7 consists of cullin-7, RBX1, adaptor protein Skp1, and the substrate receptor FBXW8.
Over the past 13 years, a number of transgenic (Tg) and knockout (KO) mouse models have been established for key components of the SCF E3 complexes. Here, we provide a comprehensive review of the existing Tg and KO mouse models of the SCF E3 components, and further discuss how these models have led to our current understanding of physiological functions of SCF E3s and their mechanisms of action. We will end with a discussion of future directions of this research area, and the potential applications of these mouse models towards understanding various human diseases, particularly cancer, which is often associated with alterations of SCF E3 ubiquitin ligases33.
S-phase kinase-associated protein 1 (SKP1)
SKP1 was first identified in 1995 as one of two essential elements of the cyclin A-CDK2 S-phase kinase34, and later found to promote ubiquitylation and degradation of various cell cycle regulators20. SKP1 functions as an adaptor protein that links Cul-1/Cdc53 and the F-box protein to form the SKP1-cullin 1-F-box protein ubiquitin ligases15,17. In yeast, however, several non-SCF functions of Skp1 have been reported with Skp1 involvement in recycling of internalized proteins35, and in regulation of cytokinesis and cell division36.
At the present time, no Skp1 KO mouse model is available. One group, however, generated Tg mice expressing a Cul-1 deletion mutant (Cul1-N252), which is capable of sequestrating and inactivating Skp137. When expressed in the T-cell lineage, Cul1-N252 causes hypoplasia and reduced proliferation in the lymphoid organs. After a latent period, however, the Cul1-N252 Tg mice develop T-cell lymphomas with high penetrance. Importantly, T-cell depletion and subsequent lymphoma formation can be largely rescued in Cul1-N252, Skp1 double Tg mice, indicating that sequestration and inactivation of endogenous Skp1 has a causal role37. Furthermore, lymphomas developed in Cul1-N252 Tg mice showed remarkable karyotype heterogeneity associated with c-Myc overexpression. These results suggest a key role for Skp1 in the maintenance of genomic stability and euploidy to prevent neoplastic transformation37. Interestingly, a recent study showed that expression of SKP1A (p19, S-phase kinase-associated protein 1A) is significantly reduced in sporadic Parkinson's disease38. RNAi-mediated silencing of SKP1A in neuronal cells increases susceptibility to cell death, whereas intranigral stereotaxic injection of a lentiviral vector targeting SKP1A induces pathological and behavioral deficits in mice38. Thus, establishment of a conditional Skp1 KO mouse model with targeted neuronal deletion of Skp1 may provide an in vivo animal model to study this type of Parkinson's disease.
Cullins
The cullin family of proteins was first identified in 1996 as being required for cell cycle exit in C. elegans39 and for the G1-to-S-phase transition in budding yeast40. The family members are characterized by an evolutionarily conserved cullin-homology domain, and serve as molecular scaffolds to facilitate the assembly of multimeric CRLs with SCF/CRL-1 as the founding member. Activity of CRLs requires conjugation of cullins with the ubiquitin-like molecule, NEDD8, a process known as cullin neddylation, which induces a conformational change that increases the proximity between the E2 enzyme and the substrates, and prevents the inhibitory association of CAND1 (cullin-associated neddylation-dissociated protein 1)41,42. Mammalian cullin protein family has eight members, including cullin-1, -2, -3, -4A, -4B, -5, -7, and -916. Although among the 8 cullins, only cullin-1 is a component of the SCF E3 ligases, this review will cover all cullins, with a focus on those for which mouse KO models are available.
Cullin-1
Cul-1 is the most extensively characterized member of the cullin family and a key component of SCF E3s (Figure 2A). To understand the physiological roles of Cul-1 in mammalian cells, mouse KO studies were performed in 1999. Cul-1 KO causes early embryonic lethality at E6.5 before the onset of gastrulation. Accumulation of cyclin E proteins, but not cyclin E mRNA, was found in Cul-1−/− embryos, blastocytes, and mouse embryonic fibroblasts (MEFs), along with increased p53 levels and apoptosis in the embryonic ectoderm43,44. Thus, Cul-1 has an indispensable role in both cell cycle progression and early embryogenesis, and cyclin E is a physiological substrate of SCF E3. Unfortunately, neither study conducted the rescue experiment by simultaneous deletion of cyclin E to determine whether accumulated cyclin E is causally related to the embryonic lethality phenotype. Nevertheless, the embryonic lethal phenotype of Cul- 1 total KO calls for the necessity of a future conditional KO mouse model to study the organ-specific role of Cul-1 in development and diseases such as cancer, as Cul-1 is overexpressed in 40% of lung cancers45, as well as during human melanomagenesis46.
Cullin-3
Cul-3 forms a complex with RBX1 and BTB (bric-a-brac, tramtrack, broad-complex domain) proteins (as substrate receptors) to constitute an active CRL-316 (Figure 2C). Interestingly, Cul-3, but not Cul-1, binds to free cyclin E in mammalian cells and is important for the ubiquitylation and degradation of free cyclin E47. More importantly, this study generated a Cul-3 KO mouse model to further define the in vivo physiological role of Cul-3. Like Cul-1 KO, Cul-3 KO in mice also causes an early embryonic lethal phenotype at E6.5–7.5 with marked accumulation of cyclin E proteins in the extraembryonic ectoderm and in the trophectoderm. Cul-3−/− cells in the extraembryonic ectoderm undergo a standard mitotic cell cycle, whereas Cul-3−/− cells in the trophectoderm are blocked from S-phase entry, indicating a cell-type-specific effect of Cul-3 on S-phase regulation47. Again, this study demonstrated that Cul-3 is required for mouse embryogenesis and that cyclin E is a physiological substrate of Cul-3-based E3 ligase. However, the causal relationship between cyclin E accumulation and early embryonic lethality was not determined by an in vivo rescue experiment.
Several Cul-3 conditional KO mouse models have been established to further define the relationship between Cul-3 and cyclin E as well as the organ-specific role of Cul-3. Cul-3−/− MEFs show increased cyclin E levels and reduced cell viability. Liver-specific deletion of Cul-3 causes a dramatic increase in cyclin E levels, along with an increase in cell size and ploidy, indicating that Cul-3-based E3 ligases promote the constitutive degradation of cyclin E, which is essential for the maintenance of the quiescent status of mammalian cells48. In a more recent study, the same group revealed a potential role of Cul-3 in liver progenitor and stem cells49. The liver-specific deletion of Cul-3 causes a persistent and massive expansion of hepatic progenitor cells, which upon differentiation undergo remarkable DNA damage, thereby leading to a cellular senescence response and cell death. This process appears to be controlled by p53, as simultaneous loss of Cul-3 and p53 in hepatic progenitors transforms these cells to highly malignant tumor-initiating cells that form largely undifferentiated tumors in nude mice. More significantly, loss of Cul-3 and p53 results in the formation of primary hepatocellular carcinomas in these mice. Thus, Cul-3 expression during hepatic differentiation appears to have a safeguard role against the formation of tumor-prone progenitor cells49. Consistent with this notion, loss of Cul-3 expression was observed in a large portion of human liver cancers and directly correlated with tumor dedifferentiation49. Furthermore, a recent study revealed that Cul-3 binds to the promyelocytic leukemia zinc finger (PLZF) transcription factor and BCL6 transcription factor, respectively50. The T cell-specific (driven by CD4-Cre) or B cell-specific (driven by CD19-Cre) Cul-3 KO studies showed that Cul-3 likely directs the differentiation of several T- and B-cell effector programs and may be involved in the oncogenic role of PLZF and BCL6 in the development of leukemias and lymphomas, respectively50. Finally, it is important to note that in human cancers the Cul-3 adaptor, Keap1, is frequently mutated, and its two well-characterized substrates, Cyclin E and Nrf2, are often overexpressed or activated by mutations that disrupt Cul-3-dependent regulation, further arguing for a critical role of the CRL-3 E3 ligase in modulating tumorigenesis51,52,53. Given the recent data that Cul-3-KLHL20 is associated with prostate cancer progression54 and Cul-3 overexpression is correlated with breast cancer progression55, and that Cul-3 mutations are detected in squamous cell lung cancers56 as well as cause hypertension and electrolyte abnormalities in humans57, the tissue-specific Cul-3 KO mouse models would be useful to mechanistically study these human diseases.
Cullin-2 and cullin-5
Cul-2 forms a complex with RBX1, adaptor proteins Elongin B/C and the substrate receptor protein von Hippel-Lindau (VHL) to construct an active CRL-2 E3 with HIF-1α58,59 as its best known substrate (Figure 2B), whereas Cul-5 forms a complex with RBX2/SAG, adaptor proteins Elongin B/C, and receptor proteins SOCS (suppressors of cytokine signaling) to form an active CRL-5 E316 (Figure 2E). No KO mouse models are currently available for these two cullins to understand their in vivo physiological functions. However, it is anticipated that total KO of either Cul-2 or Cul-5 may cause embryonic lethality, given the fact that CRL-2 and CRL-5 promote, respectively, the ubiquitylation and degradation of important proteins such as HIF-1α58,59 or SRC60 along with many other substrates (for reviews, see6,61,62). Nevertheless, conditional tissue-specific KO mouse models should be generated for these two cullins to study their organ-specific functions. The Cul-2 conditional KO mouse model may also be useful to study colon tumorigenesis, as somatic mutations of Cul-2 were detected in a fraction of human colon cancers63.
Cullin-4A and cullin-4B
In mammalian cells, Cul-4 is encoded by two closely related genes, Cul-4A and Cul-4B, which share about 82% sequence identity with redundant functions64,65. Cul-4A or Cul-4B can each form a complex with the adaptor protein DDB1 (DNA damage-binding protein 1) and the substrate receptor protein DCAF (DDB1-CUL-4-associated factor) to constitute an active CRL-4A or CRL-4B E3 (Figure 2D). An early Cul-4a KO study using the Cre/loxP strategy to ablate exon 1 of the Cul-4a gene showed that Cul-4a deletion in Cul-4b wild-type background causes embryonic lethality at E7.5, suggesting that Cul-4a is essential to early embryogenesis, and that Cul-4a and Cul-4b are functionally non-redundant66. However, a later study by another group showed that germline deletion of exons 17-19 of Cul-4a (deletion of the cullin homology domain, thus abrogating Rbx1 binding) has no effect on embryonic development. Mice were born alive and healthy without developmental abnormalities65. The discrepancy between these two studies can be explained by the inadvertent ablation of a 529-base pair region upstream of the first exon of the Pcid2 gene that resides on the complementary strand adjacent to Cul-4a exon 1 and encodes an uncharacterized protein with homology to essential proteasome subunits65. The later study also showed that skin-specific deletion of Cul-4a causes remarkably enhanced resistance to UVB-induced skin carcinogenesis. This is likely attributable to the accumulation, upon Cul-4a deletion, of both DDB2 and XPC DNA-damage sensors, as well as the p21 damage checkpoint effector, leading to an enhanced activity of the global genomic repair pathway and reinforced UV-responsive G1 DNA-damage checkpoint65. Thus, although Cul-4a and Cul-4b appear to be functionally redundant during embryogenesis, Cul-4a does play a critical role in establishing the cellular DNA repair threshold, a function not shared by Cul-4b. Furthermore, Cul-4a appears to regulate both cell proliferation and genome stability, as demonstrated by yet another study in which Cul-4a was inactivated via targeted deletion of its exons 4-8 (encoding the N-terminal DDB1-binding domain)67. In that study, Cul-4a−/− cells were found to exhibit deregulated p53 and p27 expression with severe deficiency in cell proliferation and signs of genomic instability67, although Cul-4a−/− mice are alive and healthy64. Finally, two recent studies showed that Cul-4a deletion (at exons 17-19 or 4-8) affects male (but not female) fertility and causes severe deficiencies in spermatogenesis with increased apoptosis in the testes, which is associated with elevated levels of phospho-p53 and Cdt168,69. Yin et al.69 showed that the reproductive defects in Cul-4a−/− males largely resulted from the distinct and non-overlapping expression patterns of the two Cul-4 genes in different stages, which led to the meiotic arrest at the pachytene to diplotene stage and defects in resolving meiotic chromosomal crossover in spermatocytes. These studies revealed a possible indispensable function for Cul-4a during the meiosis of male germ cells.
Most recently, two groups reported Cul-4b KO studies70,71. One study with targeted deletion of the exons 3-5 of Cul-4b showed that Cul-4b−/− embryos exhibit severe developmental arrest and die before E9.5 with cyclin E accumulation70. Even heterozygous deletion of Cul-4b resulted in severe developmental delay with disorganized vascularization in placentas. The authors concluded that Cul-4b is indispensable for embryonic development in mouse70. In another study, the exons 4-5 of Cul-4b, which encode the DDB1-binding domain of Cul-4b, were targeted71. With the chicken-actin, Cre, which causes Cul-4b deletion in both the embryo proper and extra-embryonic tissues, the authors observed embryonic lethality at E7.5. However, when Cul-4b was deleted in the epiblast using the Sox2-Cre (targeted Cul-4b deletion in the embryo proper only), viable Cul-4b−/− mice were produced likely due to the possible compensation effect of Cul-4a. Thus, Cul-4b is essential for the development of extra-embryonic tissues, which lack Cul-4a expression, but is dispensable for embryogenesis itself71. Given the fact that the Cul-4A gene is amplified or overexpressed in a number of human cancers (for review see61,72), which is associated with poor prognosis for cancer patients73, whereas Cul-4B mutations are associated with human X-linked mental retardation (XLMR) syndrome74,75, these Cul-4 conditional mouse models will be very useful for cancer research (Cul-4a) as well as for the study of neuronal and behavioral deficiencies seen in human Cul-4B XLMR patients (Cul-4b)76. Along this line, a conditional Cre-dependent gain-of-function Cul-4a Tg mouse model has been established77, which can be particularly useful for determining a potential causal role of Cul-4a overexpression in diseases, as seen in many human cancers61,72, especially in organ-specific tumorigenesis.
Cullin-7 and cullin-9/PARC
Cul-7 forms a complex with the adaptor protein SKP1, RBX1, and the receptor protein FBXW8 to constitute an active CRL-7 (Figure 2F), whereas very little is known about other components that may bind to Cul-9/Parc to form an active CRL-916. Cul-7−/− mice die immediately after birth due to respiratory distress. Vascular defects are seen in both the embryo and placenta, indicating a vital role for Cul-7 in cell differentiation and vascular morphogenesis78. On the other hand, Cul-9/Parc KO mice are born at the expected Mendelian ratios and show no apparent phenotype, suggesting that the Cul-9 gene is dispensable for embryonic development79. As Cul-7 mutations have been found in human 3-M syndrome, an autosomal recessive condition characterized by severe pre- and post-natal growth retardation80,81, and short stature syndrome82, as well as in clear cell renal cell carcinomas83, conditional Cul-7 KO mouse models, if generated, may be useful for studying these diseases.
Figure 3 summarizes the available KO models of cullin family members with a brief description of the KO phenotype and accumulated substrates, which likely contribute to the exhibited phenotypes.

Major physiological functions of cullins revealed by genetically engineered mouse models. Among the cullin family members, KO mouse models have been generated for Cul-1, Cul-3, Cul-4, Cul-7, and Cul-9, but not yet for Cul-2 and Cul-5. Functional consequences of the total or conditional KO of cullins are shown along with the involved substrates and pathways.
F-box proteins
In the human genome, there are ∼69 F-box proteins84, which have been classified into three categories: (1) WD40 domain-containing (FBXWs), (2) LRRs-containing (FBXLs), and (3) other diverse domains-containing (FBXOs)85,86. It is well-established that the F-box proteins determine the substrate specificity of the SCF E3s and that a substrate generally has to be phosphorylated by a kinase(s) prior to being recognized by an F-box protein for targeted ubiquitylation and degradation15,17, with one known exception in which substrates are glycosylated before being recognized by Fbx287. To date, only a small fraction of the 69 F-box proteins has been characterized with at least one corresponding substrate identified. The list includes β-TrCP1 (β-transducin repeat-containing protein) (also known as FBXW-1), β-TrCP2 (also known as FBXW-11), FBXW-2, -5, -7, and -8; SKP2 (also known as FBXL-1), FBXL-3, -5, -6, -12, -20, and -21; and FBXO-2, -3, -4, -6, -7, -8, -31, -32, and -3388,89,90. In this review, we will mainly focus on the three best characterized mammalian F-box proteins, namely β-TrCP, SKP2, and FBXW7, and discuss their in vivo physiological functions revealed through the studies of their respective Tg and/or KO mouse models (Figure 4). Available mouse models for other F-box proteins will also be described briefly.

Major physiological functions of three well-known F-box proteins revealed by genetically modified mouse models. The SCF E3 consists of SKP1, Cul-1, RBX1/SAG and one of 69 F-box proteins that serve as receptors for substrates and thereby determine substrate specificity. Among the 69 F-box proteins, β-TrCP, SKP2, and FBXW7 are well-characterized in vivo using genetically modified mouse models. Shown are selective substrates that contribute to the KO/Tg phenotypes and altered biological processes.
β-transducin repeat-containing protein (β-TrCP)
Human β-TrCP, first identified in 1998 as a binding partner of HIV-1 Vpu protein in a yeast two-hybrid screening91, has two family members, namely β-TrCP1 (also known as FBXW1, FBW1A, and FWD1) and β-TrCP2 (also known as FBXW11, FBW11, FBXW1B, FBX1B, and HOS). Human β-TrCP1 and β-TrCP2 are highly evolutionarily conserved as shown by the comparison with their counterparts found in Xenopus92 and Drosophila (known as Slimb)93. The β-TrCP family member contains a 42-48 amino acid F-box motif at the N-terminus for binding to other components of the SCF complex and seven WD40 repeats at the C-terminus for recognition of substrates, which normally contain the conserved DSGxxS motif94. Two typical substrates of β-TrCP1/2 are IκBα and β-catenin, which are recognized after phosphorylation of both serine residues of the DSGxxS motif (for reviews see88,94). β-TrCP mainly acts as an oncoprotein, but in some situations as a tumor suppressor, depending on the function of the targeted substrates (for reviews, see2,31,88,94) (Figure 4). Although β-TrCP1 and β-TrCP2 are biochemically indistinguishable in vitro, it remained unclear before the KO studies whether the two β-TrCP paralogues have overlapping functions in vivo or whether each of them recognizes a unique set of substrates.
Two studies on total β-Trcp1 KO were reported in 2003 from two independent laboratories. In the first study, β-Trcp1 was disrupted by targeted deletion of exon 1 (including the translation-initiation site). The β-Trcp1−/− mice are viable and fertile without any observed abnormality up to 16 months95. However, β-Trcp1−/− cells (lymphocytes and MEFs) show reduced degradation of IκB proteins (IκBα and IκBβ) with corresponding inhibition of NF-κB activity. β-Trcp1 deletion also causes accumulation of nuclear, but not cytoplasmic, β-catenin in MEFs. Biologically, β-Trcp1 deletion results in reduced rate of cell proliferation, with increased polyploidy95 and abnormal retinal development96. In the second study, β-Trcp1 was disrupted with targeted deletion of exon 5 (encoding the F-box domain)97. These β-Trcp1−/− mice are again viable, but show reduced fertility. The corresponding β-Trcp1−/− MEFs display mitotic abnormalities with centrosome overduplication and chromosome misalignment. Among known substrates of β-Trcp1, accumulation was seen for cyclin A, cyclin B, and Emi1, but surprisingly not IκBα and β-catenin, whose accumulation requires additional silencing of β-Trcp2 by siRNA97. Accumulation of Emi1 appears to contribute to the moderate impairment in spermatogenesis and male fertility97. These studies clearly suggest that β-Trcp1 and β-Trcp2 are functionally redundant during embryonic development as well as in controlling the stability of IκBα and β-catenin in vivo. To gain a better understanding of β-Trcp redundancy, Kanarek et al.98 recently generated a mouse strain with inducible β-Trcp2 knockdown in combination with β-Trcp1 KO (designated as KO1/KD2). Even though the β-Trcp2 level is reduced to only ∼10% of the basal level in most tissues upon tetracycline treatment, the KO1/KD2 mice are still viable and healthy, except for a severe testicular defect. The defect is fully reversible upon β-Trcp2 restoration and is caused apparently by the accumulation of a single β-Trcp substrate, Snail1, as additional depletion of Snail1 completely restored spermatogenesis98. Detailed analyses performed in this study also suggest a non-redundant role of β-Trcp1 and β-Trcp2 in spermatogenesis.
Although overexpression and mutations of β-TrCP1 were reported in multiple human cancers (for reviews, see2,31,88,94), the causal effect of β-Trcp on tumorigenesis was best demonstrated using Tg mouse models. Whereas mammary glands of β-Trcp1−/− female mice display a hypoplastic phenotype, targeted expression of β-Trcp1 by the MMTV promoter in mammary epithelia of female mice causes enhanced proliferation with increased NF-κB activity. About 38% of Tg mice develop tumors, including mammary, ovarian, and uterine carcinomas. In contrast, Tg expression of β-Trcp1 in lymphoid organs produces no phenotype, indicating that the oncogenic activity of β-Trcp1 is tissue-specific99. In a separate study, Tg expression of β-TrCP1 or β-TrCPΔF (a dominant-negative mutant with the F-box deleted) in intestine, liver, and kidney induces intestinal adenomas as well as hepatic and urothelial tumors, likely through nuclear accumulation of β-catenin100.
S-phase kinase-associated protein 2 (SKP2)
SKP2 was first identified in 1995 as the second essential element of the cyclin A-CDK2 S-phase kinase34, and later found to promote ubiquitylation and degradation of cell cycle regulators20. Many follow-up cell culture studies showed that Skp2 promotes proliferation and survival of cancer cells in part through targeted degradation of various regulatory substrates, particularly tumor suppressors, such as p21, p27, and Foxo1 (for reviews see2,31,88) (Figure 4).
To define the in vivo physiological function of Skp2, a Skp2 total KO mouse model was generated by deletion of exons 1-4 that removes the region from the translation-initiation site to the site corresponding to the C-terminus of the F-box domain101. Skp2−/− mice are viable and fertile, but smaller in size. Skp2−/− MEFs, as well as cells from multiple organs, including liver, lung, kidney, and testis have enlarged nuclei with polyploidy and centrosome overduplication. Furthermore, Skp2−/− MEFs show reduced growth rates and increased apoptosis with accumulation of p27 and cyclin E, indicating that p27 and cyclin E are in vivo substrates of Skp2101. To pursue a causal role of p27 accumulation in the observed phenotype, two independent groups performed in vivo rescue experiments102,103. The results showed that simultaneous deletion of p27 rescues the defects in cell size and ploidy in Skp2−/− mice, as well as defects in cell cycle progression and centrosome overduplication in Skp2−/− cells102,103. These rescue experiments demonstrate that p27 is a major physiological target of SCFSkp2, whereas accumulation of cyclin E alone is not responsible for the cellular abnormalities of Skp2−/− mice.
The Skp2 KO mouse model has also revealed the involvement of Skp2 in the following biological processes and/or disease conditions. First, Skp2 regulates the quiescence maintenance and self-renewal of hematopoietic stem cells (HSCs). Skp2−/− HSCs show reduced quiescence and populate faster than wild-type control cells through accelerated cell cycle entry due to increased cyclin D1 expression. Moreover, Skp2 loss rescues the defect in long-term reconstitution ability of HSCs caused by Pten inactivation and sensitizes HSCs to chemotherapy104. Second, Skp2 is required for ovarian development. Skp2−/− female mice show severely compromised gamete development from the embryonic stage to follicular growth in the adult ovary, eventually leading to a decreased functional gamete reserve and reduced fertility. The defect is caused by p27 accumulation, which can be rescued by the simultaneous deletion of p27105. Third, Skp2 is also required for proliferation of vascular smooth muscle cells. In Skp2−/− mice, neointima formation is retarded compared to wild-type littermates after carotid ligation106. Fourth, Skp2 controls obesity. Skp2−/− mice had a 50% reduction in both the subcutaneous and visceral fat pad mass and adipocyte number, which can be completely rescued by p27 deletion107. Furthermore, Skp2−/− mice are more resistant to the development of obesity and obesity-related insulin resistance induced by a high-fat diet or by the lethal yellow agouti mutation, likely due to inhibition of the increase in adipocyte number108. Finally, Skp2−/− mice are also more resistant to nephropathy triggered by unilateral ureteral obstruction (UUO), a situation that induces Skp2 expression109. Moreover, this resistance is abrogated by the simultaneous deletion of p27, indicating that p27 accumulation has a causal role in the suppression of UUO renal injury110.
Cell culture studies showed that Skp2 typically acts as an oncogene by promoting the degradation of many tumor suppressors, particularly p27, whereas human tissue studies showed Skp2 overexpression in many human cancers that is directly associated with poor patient prognosis (for reviews, see2,31,88). The causal relationship between Skp2 and tumorigenesis has been extensively examined using both KO and Tg mouse models. A recent in vivo study links Skp2 to cellular senescence induced by oncogenic signals or inactivation of tumor suppressors. In response to inactivation of tumor suppressors (Arf, Pten), Skp2 deletion triggers a cellular senescence response in a manner independent of p19Arf-p53, but dependent on Atf4/p21/p27. Consequently, Skp2 deletion reduced the rate of sarcoma/lymphoma formation and extended the lifespan of Arf−/− mice. Consistently, Skp2 deletion also inhibits prostate tumorigenesis triggered by prostate-specific Pten loss and extends the life-span of these mice111, and attenuates BCR-ABL-induced myeloproliferative disease112. These studies clearly demonstrate that Skp2 is an oncogene or onco-cooperating gene, and is a promising target for cancer prevention and treatment. In another study, Skp2 ablation in Rb1+/− mice blocks spontaneous tumorigenesis in a p27-dependent manner, but fails to inhibit tumorigenesis induced by a chemical carcinogen, ENU113. However, Skp2 deletion in mice does not significantly inhibit Myc-driven proliferation and lymphomagenesis114, nor does it affect Myc-mediated tumorigenesis in the oral epithelium115. The role of Skp2 in proliferation and tumorigenesis is further demonstrated in several Tg mouse models. Skp2, when targeted to express in the T-lymphoid lineage, along with activated N-Ras, promotes N-Ras-induced T-cell lymphoma formation with shorter latency, higher penetrance, and significantly decreased survival, indicating a strong N-Ras-cooperative effect116. Moreover, prostate-specific expression of Skp2 downregulates p27 and consequently induces hyperplasia, dysplasia, and low-grade carcinoma117. Taken together, Skp2 appears to be a tissue-specific oncogene or onco-cooperative gene whose deletion suppresses tumorigenesis.
F-box and WD-40 domain protein 7 (Fbxw7)
The first member in the gene family of FBXW7 (also known as Fbw7, Sel-10, hCdc4, or hAgo) was identified in 1973 in budding yeast by Hartwell et al.118, designated as Cdc4118. Human FBXW7 is a well-characterized F-box protein that binds to its substrates through a Cdc4 phosphodegron (CPD) consensus motif, IL-I/L/P-pT-P, that is present on the substrates targeted for proteasomal degradation119. Almost all FBXW7 substrates are oncogenic proteins, including c-Myc, c-Jun, cyclin E, and Notch (for review, see119) with one exception which is the tumor suppressor neurofibromatosis type 1 (NF1), a physiological substrate of FBXW7 newly identified in our laboratory120. Functionally, FBXW7 is a haploinsufficient tumor suppressor121 with mutations found in many human cancers (for review, see119).
The in vivo physiological functions of Fbxw7 during embryonic development were characterized by two KO studies. Fbxw7 total KO causes embryonic lethality; Fbxw7−/− embryos die in utero around E10.5–11.5 with remarkable abnormalities in vascular development in the brain and yolk sac, as well as deficient heart chamber maturation122,123. These abnormalities are likely associated with the dysregulation of Notch signaling, along with cyclin E accumulation122,123, but no genetic rescue experiments were conducted to firmly establish the causal relationship. A subsequent study showed that Fbxw7 is a haploinsufficient tumor suppressor gene, as Fbxw7+/− mice display a greater susceptibility to radiation-induced tumorigenesis with the wild-type allele being retained and expressed121. Fbxw7+/− mice in the p53+/− background develop a wide range of tumors in epithelial tissue, such as the lung, liver, and ovary. MEFs from Fbxw7-deficient mice or with Fbxw7 knockdown have higher levels of Aurora-A kinase, c-Jun, and Notch4, but not of cyclin E. It was proposed that Fbxw7 acts as a tumor suppressor by maintaining genomic stability121.
Studies with lineage- and tissue-specific KO of Fbxw7 have revealed its diverse functions. Conditional KO mice with ablation of Fbxw7 in the T-cell lineage showed that Fbxw7 deficiency in immature T cells causes a failure in cell cycle exit, leading to thymic hyperplasia and subsequent development of thymic lymphoma, in part due to the excessive accumulation of c-Myc124. Mature T cells from Fbxw7−/− mice, however, fail to proliferate in response to mitogenic stimulation, but instead undergo apoptosis, which is associated with the accumulation of c-Myc and p53, and can be rescued by the additional deletion of p53. Thus, Fbxw7 regulates the cell cycle in a manner dependent on the differentiation stage124. Targeted deletion of Fbxw7 in HSCs results in (a) defective quiescence maintenance, impaired self-renewal, and loss of competitive repopulating capacity in one study125, and (b) premature depletion of HSCs and p53-dependent apoptosis in the other report126. Furthermore, Fbw7 deletion confers a selective advantage to cells with suppressed p53 function, leading to the development of T-cell acute lymphoblastic leukemia126. How Fbxw7-deficient cells evade cell death in the setting of elevated accumulation of c-Myc remains elusive, but it is likely due to the accumulation of Mcl-1, a prosurvival protein which has recently been characterized as a novel substrate of Fbxw7127,128. Indeed, increased Mcl-1 levels were found in thymus tissues, as well as the resulting thymic lymphoma and acute lymphoblastic leukemia cells from mice with the T-cell lineage-specific deletion of Fbxw7124,127.
Other conditional KO studies showed unique functions of Fbxw7 in various organs. For example, Fbxw7 is haploinsufficient for intestinal tumor suppression in the conditional gut-specific Fbxw7 KO mice129. Liver-specific Fbxw7 ablation induces abnormalities in lipid metabolism and cell differentiation, likely due to the accumulation of SREBPs and Notch1, respectively130. Additionally, mice with conditional brain-specific deletion of Fbxw7 exhibit severely impaired stem cell differentiation and increased progenitor cell death. These mice die shortly after birth with morphological abnormalities of the brain and the absence of suckling behavior131,132. Thus, Fbxw7 serves as a key regulator of progenitor cell viability and differentiation of neural stem cells in the brain, which is associated with its regulation of c-Jun and Notch (Notch1 and Notch 3), respectively131,132. It is likely that the tissue-specific function of Fbxw7 is mediated by selective targeting of unique sets of Fbxw7 substrates in tissue- and cell context-dependent, and spatially and temporally controlled manners. Most importantly, Fbxw7 alterations have been observed in many human cancers (for review, see133), consistent with the finding that it is a haploinsufficient tumor suppressor in mice.
Other F-box proteins
(1) Fbxw8 (also known as FBW6, FBW8, FBX29, FBXW6, or FBXO29). Fbxw8 forms a complex with Cul-7-RBX1-SKP1 to promote degradation of the insulin receptor substrate 1134 and cyclin D1135, thus regulating cell proliferation and survival. Two Fbxw8 total KO studies were reported. In the first study, exon 2 that encodes the F-box domain was disrupted. About two-thirds of Fbxw8−/− embryos die in utero, whereas the remaining one-third are born alive and grow to the adulthood. Phenotypically, Fbxw8−/− embryos show intrauterine growth retardation and abnormal development of the placenta136. In the second study, a gene-trap approach was used with the targeting vector inserted at the 3′ end of exon 3 of the Fbxw8. Again, about 30% of Fbxw8−/− mice survived the birth, but these mice remained smaller than their wild-type littermates in both body weight and organ sizes137. Thus, Fbxw8 appears to have a significant role in growth control.
(2) Fbx4 (also known as Fbxo4). Fbx4 binds to αB crystallin to assemble an active SCFFBX4-αB crystallin E3 for targeted degradation of cyclin D1, and thus plays inportant roles in inhibiting cell cycle progression138, suppressing oncogenic transformation139, and maintaining genomic stability140. Fbx4−/− mice were generated to investigate the in vivo tumor suppressor function of Fbx4. Whereas Fbx4−/− mice are viable without major developmental defects, Fbx4−/− MEFs do exhibit cyclin D1 accumulation and nuclear localization with increased susceptibility to Ras-induced transformation. More importantly, both Fbx4+/− and Fbx4−/− mice develop multiple types of tumors, including lymphomas, histiocytic sarcomas and, less frequently, mammary and hepatocellular carcinomas. Finally, a majority of Fbx4 mutations identified in human esophageal tumors are hemizygous and occur in the N-terminal regulatory region of FBX4, which disrupt ligase dimerization, suggesting that a reduction in the Fbx4 dosage and SCFFbx4 activity is sufficient to stabilize cyclin D1 and subsequently trigger neoplastic growth139. Thus, Fbx4 is a bona fide tumor suppressor139,141.
(3) Fbx2 (also known as Fbxo2). Fbx2 binds specifically to proteins attached to N-linked high-mannose oligosaccharides, and targets N-glycosylated proteins for ubiquitylation and degradation87. A recent study showed that Fbx2 promotes the degradation of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1), leading to the attenuation of Alzheimer's disease amyloidosis and improvement of synaptic function142. Mice with targeted deletion of Fbx2 develop accelerated, age-related hearing loss starting at 2 months. Cellular degeneration begins in the epithelial support cells of the organ of Corti, whereas progressive degeneration is observed in hair cells and the spiral ganglion, but not in the brain itself. This study demonstrates that Fbx2 is essential for inner ear homeostasis and acts to prevent age-related hearing loss143. The relationship between the hearing loss and potential accumulation of BACE1 or other N-glycosylated proteins, however, still remains unclear.
(4) Fbxo7 (also known as Fbx7). An early study showed that Fbxo7 promotes the degradation of hepatoma upregulated protein (HURP)144. A Tg mouse model was established with a LacZ insertion into the Fbxo7 locus to reduce its expression. Homozygous Fbxo7LacZ mice showed significantly increased pro-B cell and pro-erythroblast populations, consistent with a role of Fbxo7 in inhibiting proliferation and/or promoting maturation of hematopoietic precursor cells145. It is, however, unknown whether these phenotypic changes have anything to do with HURP.
(5) Fbxl5. FBXL5 contains a unique N-terminal hemerythrin-like domain that directly binds to iron. Under iron-depleted conditions, FBXL5 is degraded in a manner dependent on this iron-binding domain. Under iron-replete conditions, however, FBXL5 forms an active E3 with SKP1-CUL1, and catalyzes the ubiquitylation and degradation of iron regulatory protein 2 (IRP2). Thus, FBXL5 regulates intracellular iron homeostatsis146. This function was further confirmed by a recent mouse KO study147. Fbxl5−/− mice die in utero with excessive accumulation of iron and IRP2, which is rescued by Irp2 ablation. Liver-specific deletion of Fbxl5 results in deregulation of both hepatic and systemic iron homeostasis, and eventually leads to steatohepatitis. Thus, Fbxl5 plays a major role in controlling an appropriate supply of iron to cells via modulating IRP2 levels147.
(6) Fbxo45. Fbxo45 is an F-box protein with restricted expression in the nervous system. It fails to bind to Cul-1 due to an amino acid substitution at the consensus Cul-1 binding site, but specifically associates with protein associated with Myc (PAM), another RING finger-type ubiquitin ligase, through its SPRY domain148. Fbxo45−/− mice die soon after birth due to respiratory distress, and Fbxo45−/− embryos show abnormal neural development148. Similar defects are also found in mice lacking Phr1 (the mouse ortholog of PAM), indicating that Fbxo45 has an important role in neural development possibly by forming a novel Fbxo45-PAM ubiquitin ligase complex. Although Fbxo45 promotes p73 degradation149, the potential involvement of p73 in its biological function is unknown.
(7) Fbx15. Fbx15 is a downstream target of the transcription factor Oct3/4 which is essential for self-renewal of ES cells. Homozygous deletion of Fbx15 has no effect on embryogenesis in mouse, nor does it affect the cellular morphology or capacity for proliferation and differentiation of mouse embryonic stem (ES) cells, indicating that Fbx15 is not an essential gene for embryonic development150.
The RING components, RBX1, and RBX2/SAG
The RING component of SCF E3 ubiquitin ligases is composed of two family members, RBX1 and RBX225,72. RBX1, also known as ROC1 or HRT1, was initially cloned in 1999 in four different laboratories21,22,23,24. RBX1 consists of five exons and four introns, and encodes 108 amino acids with a molecular weight of about 14 kDa in humans21,22,23,24. SAG, also known as RBX2 or ROC2, was originally cloned in our laboratory as a redox-inducible antioxidant protein25, containing four exons and three introns with exon 2 spliced out to give rise to an open reading frame of 113 amino acids151,152. Both RBX1/ROC1 and SAG/RBX2/ROC2 contain an evolutionarily conserved RING zinc finger domain at the C- terminus, which is required for their ubiquitin ligase activity26,153. Both RBX1/ROC1 and SAG/RBX2/ROC2 are capable of binding to six members of the cullin family (Cul1-3, Cul-4A/4B, and Cul-5) under overexpressed conditions and show an in vitro E3 ubiquitin ligase activity when forming a complex with Cul-126,154. Before our KO studies, the only known difference between RBX1 and RBX2 is that RBX1 is constitutively expressed and prefers to bind with Cul-2/VHL, whereas SAG is stress-inducible and more selectively binds to Cul-5/SOCS155,156. Our recent mouse KO studies (see below) revealed that the in vivo functions of Rbx1 and Sag are non-redundant during embryonic development with each protein potentially targeting different sets of substrates120,157.
RBX1/ROC1
RBX1 activates SCF E3 ligase activity by promoting cullin-1 neddylation to facilitate the ubiquitin transfer from E2 to substrates158,159. To understand the in vivo physiological function of RBX1, we attempted to generate an Rbx1 KO mouse model via a gene-trap approach157. We found that targeted inactivation of Rbx1 caused embryonic lethality at embryonic day E7.5 due to proliferation failure157. Our mechanistic study further revealed that Rbx1 inactivation caused significant accumulation of p27, which is normally not expressed in the E6.5 to E7.5 embryos157. It is, therefore, conceivable that the accumulated p27 would block cell cycle progression to suppress proliferation of the embryos. To determine whether p27 accumulation is causally related to the embryonic death at E7.5, we performed an in vivo rescue experiment and found that simultaneous deletion of p27 rescues the early embryonic lethality induced by Rbx1 inactivation and extends the embryonic life-span from E6.5 to E9.5, indicating that p27-mediated cell cycle inhibition partially contributes to the early lethality in Rbx1-deficient embryos157. Failure in embryonic rescue to full-term by p27 deletion indicates that p27 is no longer critical beyond E9.5, or the accumulation of other Rbx1-SCF substrates is lethal. Although previous studies using the yeast as a model revealed that RBX1 and RBX2/SAG are interchangeable in rescuing the death phenotype caused by ablation of Hrt1 (the only yeast homolog of RBX1/SAG)22,23,26, and the in vitro biochemical assays showed that RBX1 and RBX2/SAG are interchangeable in promoting protein polyubiquitylation26,160, our mouse Rbx1 KO study conducted under the Sag+/+ background provided the first demonstration that the two RING family members are not functionally redundant in vivo during mouse embryogenesis157. Although the precise underlying mechanism(s) remains elusive, it likely involves the preferential binding of Rbx1 to Cul-2 vs Rbx2/Sag to Cul-5156,161, resulting in the formation of different CRLs (CRL-2 vs CRL-5) to target unique sets of substrates in a cell context- and development stage-dependent manner. Future studies are needed to resolve this important issue62.
SAG/RBX2/ROC2
SAG/RBX2/ROC2, the second family member of the SCF RING component was first cloned in our laboratory as a redox-inducible protein25,162. Our subsequent work showed that SAG can also be induced by the tumor-promoting agent 12-O-tetradecanoylphorbol-13-acetate (TPA) via activator protein-1 (AP-1) transactivation155 and hypoxia via HIF1 transactivation163. Using a cell culture-based model, we found that SAG overexpression inhibits, whereas SAG siRNA knockdown enhances TPA-induced neoplastic transformation by targeting c-Jun, thus modulating AP1 activity155. To determine the role of SAG in skin tumorigenesis in a more physiological setting, we generated two independent Tg mouse lines with targeted SAG expression in epidermis, driven by the K14 promoter164. Although SAG Tg expression has no detectable effect on epidermis development and proliferation under unstressed conditions, it inhibits TPA-induced epidermal proliferation by targeting c-Jun for degradation164. On the other hand, in UV-induced epidermal proliferation, SAG Tg expression promotes the degradation of both c-Jun (pro-proliferation) and p27 (antiproliferation) with a net outcome of enhanced proliferation165, suggesting a more significant role of p27 in suppression of UVB-induced skin proliferation.
We next examined the effect of SAG Tg expression on skin carcinogenesis initiated by DMBA [7,12-dimethylbenz(a)-anthracene] and promoted by TPA or by UVB exposure. The DMBA/TPA two-stage model for skin squamous cell carcinoma (SCC), which recapitulates the development of human skin SCC, is the most commonly used model166. We demonstrated that during DMBA/TPA-induced skin carcinogenesis, SAG Tg expression caused an early-stage suppression of hyperplasia and tumor formation by promoting c-Jun degradation (in cooperation with Fbxw7), thereby inhibiting AP-1, but a later-stage enhancement of tumor growth by promoting IκBα degradation (in cooperation with β-TrCP), thus activating NF-κB and inhibiting apoptosis164. This stage-dependent effect of SAG led to a fewer number, but a bigger size of tumors per animal in SAG Tg mice, as compared to the non-Tg littermates164. Using the same SAG Tg mouse model, we found that SAG Tg expression promotes skin hyperplasia, but not skin tumors induced by UVB exposure. This is likely due to SAG-mediated simultaneous degradation of c-Jun (oncogenic) and p27 (tumor suppressive), leading to a moderate biological outcome165.
We further pursued physiological functions of Sag during embryogenesis by generating Sag KO mice using a gene-trap approach120. Like Rbx1 disruption, Sag disruption also caused embryonic lethality, but at the later stage of E11.5–12.5, likely due to poor vasculogenesis, defective neuronal development, and widespread apoptosis120. Mechanistic studies using mouse embryonic stem (ES) cells revealed that Sag inactivation caused the accumulation of Nf1, a well-known tumor suppressor that converts GTP-bound active Ras to GDP-bound inactive Ras167. Accumulated Nf1 inhibits Ras activity and the Ras-MAPK signaling pathway, thus preventing ES cells from undergoing endothelial differentiation in vitro to form blood island structures (the in vitro version of in vivo vasculogenesis), and inhibiting in vivo angiogenesis and proliferation in teratomas upon injection of the mutant ES cells into nude mice120. Significantly, the defect in endothelial differentiation can be fully rescued by the simultaneous deletion of even one Nf1 allele, which is sufficient to reactivate the Ras-MAPK pathway, indicating a causal effect of Nf1 accumulation. Indeed, detailed biochemical characterization revealed that SAG, in cooperation with FBXW7, promotes NF1 ubiquitylation and degradation; thus NF1 is a novel substrate of the SAG-SCFFBXW7 E3 ligase120.
We further determined whether the simultaneous deletion of Nf1 would rescue vascular defects in Sag−/− embryos and found that Sag−/−;Nf1−/− embryos indeed show an improved vasculature with reappearance of some trunk blood vessels as well as reduced number of apoptotic cells in the head areas, indicating a partial rescue of defects in vasculogenesis and apoptosis120. However, simultaneous Nf1 deletion failed to extend the embryonic life-span of Sag−/− embryos, indicating that Sag substrates other than Nf1 might have a key role in mouse embryonic development beyond E12.5. Nevertheless, our study clearly demonstrates an in vivo physiological function of Sag in regulating vascular and neural development through targeted degradation of Nf1 to modulate the Ras-MAPK signaling pathway. Furthermore, as this Sag KO study was conducted under the Rbx1+/+ background, our study further confirmed that Sag and Rbx1 play a non-redundant roles during mouse embryogenesis.
Both our in vitro cell culture and in vivo KO studies showed that SAG is an anti-apoptotic cellular survival protein whose overexpression protects cells from apoptosis induced by various stimuli (for review, see62,72), whereas its silencing by siRNA or its deletion by gene KO induces apoptosis120,160,168. Consistently, SAG was found to be overexpressed in multiple human cancer tissues including carcinomas of the lung, colon, stomach, and liver168,169,170, and SAG overexpression correlates with poor survival of lung cancer patients170. Our unpublished data using a conditional Sag KO mouse model showed that Sag is required for lung tumorigenesis induced by KrasG12D. Thus, Sag appears to be an onco-cooperating gene whose overexpression is causally related to tumorigenesis.
The results of our in vivo Tg and KO studies, along with in vitro cell culture-based studies on RBX1 and SAG, are summarized in Figure 5. Both Rbx1 and Sag are required for mouse embryonic development and play essential roles in regulation of cell proliferation and survival as well as carcinogenesis. Inactivation of either RBX1 or SAG inhibits carcinogenesis via various mechanisms, including apoptosis, senescence, and autophagy168,171,172. Thus, RBX1/SAG and their associated SCF E3 ligases have been considered to be attractive anticancer targets2,33,72,173,174. Indeed, MLN4924, a newly discovered small molecule inhibitor of NEDD8-activating enzyme (NAE), which inactivates the SCF/CRL E3 ligases by blocking cullin neddylation, a process required for SCF/CRL E3 activity175,176, showed impressive anticancer activity by inducing apoptosis176,177,178,179,180, senescence111,181,182, and autophagy183,184, and is being developed as the first-in-class NAE inhibitor for anticancer therapy185.

The in vivo and in vitro biological functions of the two RING finger proteins, RBX1 and SAG. The RING finger proteins of SCF E3 have two family members, RBX1 and SAG/RBX2. Both are required for mouse embryonic development through targeted degradation of p27 (for Rbx1)157 and Nf1 (for Sag)120, respectively. Upon induction of RBX1 by mitogen154 and of SAG by UV, TPA or ROS72, the RING proteins recruit other components of SCF for targeted degradation of cognate substrates. SAG Tg expression modulates skin carcinogenesis induced by DMBA/TPA by inhibiting cell proliferation via targeting c-Jun/AP-1 at the early stage, but promoting tumor growth via targeting IκB to activate NFκB at the later stage164. SAG transgenic expression also regulates epidermal hyperplasia by targeting p27 and c-Jun165. RBX1 also likely modulates carcinogenesis in part via regulating apoptosis, autophagy and senescence171,172. MLN4924, a small molecule inhibitor of NAE, blocks cullin neddylation to inactivate SCFs, leading to the induction of apoptosis, autophagy and senescence179,181,183,184, thus acting as a novel class of anticancer agent.
Conclusion and future perspectives
The SCF E3 ubiquitin ligases that operate together with the upstream E1 and E2 enzymes, and the downstream 26S proteasome, have a crucial role in the maintenance of the integrity of many basic cellular processes15,17,31,32. Over the past 15 years, we have achieved a better understanding of the functional significance of the SCF E3 ligases and their associations with human diseases through extensive studies using approaches including biochemistry, cell biology, structural biology, genomics, proteomics, and genetically modified mouse models. In this review, we provide a comprehensive overview of the existing Tg and KO mouse models generated by targeting many individual components of the SCF E3 ligases, and discuss their biological functions under in vivo physiological settings, which are summarized in Figures 3,4,5. In our opinion, future functional studies of SCF E3 ligases using genetically engineered mouse models should be extended and directed to the following areas (Figure 6).

Outline of future perspectives for studies involving mouse models of the SCF E3s. Four future directions are proposed: (1) human disease-relevant mouse models for disease characterization and intervention; (2) tissue-specific and inducible KO/Tg models of the remaining components of SCF E3s for additional functional studies; (3) rescuing mouse models for mechanistic studies; and (4) compound mouse models in combination with dominant oncogenes or loss of tumor-suppressor genes for carcinogenesis studies.
First of all, efforts should be made to generate and characterize mouse models that faithfully recapitulate the pathological conditions of human diseases. Examples include Cul-4b for human XLMR syndrome74,75, Cul-7 for 3-M syndrome80,81, and Fbxo7 for Parkinsonian-Pyramidal Disease (PPD)186,187. These models would be instrumental for the study of the disease mechanisms and the development of potential therapeutic interventions.
Second, new mouse models should be established to target the remaining components of the SCF E3s, including all 69 F-box genes, for tissue-specific Tg expression (gain-of-function) and KO (loss-of-function), ideally in an inducible manner. Successful completion of this comprehensive study will not only provide us the entire picture of physiological functions of SCF E3s, but also differentiate overlapping/redundant vs unique functions between family members in a tissue-specific manner. Examples include Cul-4a vs Cul-4b, β-TrCP1 vs β-TrCP2, and Rbx1 vs Sag/Rbx2.
Third, efforts should be made to characterize the causal role and contribution of any given substrate to the phenotype(s) of SCF E3 KOs. This can be achieved through rescue experiments by simultaneous deletion of the accumulated substrates, one at a time or in combination of a few if necessary. For example, the embryonic death phenotype of Rbx1 KO can only be partially rescued by simultaneous p27 deletion. As Cdt1, which mediates DNA-damage response and cell killing, is accumulated upon Rbx1 silencing188, KO of Cdt1 in addition to Rbx1 and p27 may lead to the full rescue of embryonic lethality or further extend the embryonic life-span. Another rescue example would be to KO Noxa, a pro-apoptotic protein which is accumulated upon Sag silencing168, in addition to Sag and Nf1, to block the widespread apoptosis seen in the Sag−/− embryos120.
Finally, the majority of the individual components of SCF E3s may not be a bona fide dominant tumor suppressor or oncogene by itself, but they may cooperate with dominant oncogenes or tumor suppressors to regulate tumorigenesis. Therefore, efforts should be made to generate compound mouse models in which manipulation of an SCF E3 component is combined with an activated oncogene (such as Kras) or an inactivated tumor suppressor (such as p53 or Pten loss) to determine whether, for example, Rbx1 or Sag, which is frequently overexpressed in human cancers168,171, indeed plays a causal role or their overexpression is just the consequence of tumorigenesis. The information gained from these series of future studies would help to justify the use of SCF E3 inhibitors, such as MLN4924176, which is currently in clinical trials185,189 for the treatment of human cancers with activated SCF E3 ligases.
References
Hershko A, Ciechanover A, Varshavsky A . Basic Medical Research Award. The ubiquitin system. Nat Med 2000; 6:1073–1081.
Nalepa G, Rolfe M, Harper JW . Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov 2006; 5:596–613.
Ravid T, Hochstrasser M . Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol 2008; 9:679–690.
Hershko A, Ciechanover A . The ubiquitin system. Annu Rev Biochem 1998; 67:425–479.
Nagy V, Dikic I . Ubiquitin ligase complexes: from substrate selectivity to conjugational specificity. Biol Chem 2010; 391:163–169.
Zhao Y, Sun Y . Cullin-RING ligases (CRLs) as attractive anti-cancer targets. Curr Pharm Des 2012 Nov 2. [Epub ahead of print]
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM . The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993; 75:495–505.
Lipkowitz S, Weissman AM . RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer 2011; 11:629–643.
Chau V, Tobias JW, Bachmair A, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989; 243:1576–1583.
Skaug B, Jiang X, Chen ZJ . The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem 2009; 78:769–796.
Spence J, Gali RR, Dittmar G, et al. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell 2000; 102:67–76.
Ulrich HD, Walden H . Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol 2010; 11:479–489.
Acconcia F, Sigismund S, Polo S . Ubiquitin in trafficking: the network at work. Exp Cell Res 2009; 315:1610–1618.
Hicke L . Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2001; 2:195–201.
Deshaies RJ, Joazeiro CA . RING domain E3 ubiquitin ligases. Annu Rev Biochem 2009; 78:399–434.
Sarikas A, Hartmann T, Pan ZQ . The cullin protein family. Genome Biol 2011; 12:220.
Willems AR, Schwab M, Tyers M . A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta 2004; 1695:133–170.
Feldman RM, Correll CC, Kaplan KB, Deshaies RJ . A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 1997; 91:221–230.
Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW . F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 1997; 91:209–219.
Bai C, Sen P, Hofmann K, et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 1996; 86:263–274.
Kamura T, Conrad MN, Yan Q, Conaway RC, Conaway JW . The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev 1999; 13:2928–2933.
Ohta T, Michel JJ, Schottelius AJ, Xiong Y . ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol Cell 1999; 3:535–541.
Seol JH, Feldman RMR, Zachariae WZ, et al. Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev 1999; 13:1614–1626.
Tan P, Fuchs SY, Chen A, et al. Recruitment of a ROC1-CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of IκBα. Mol Cell 1999; 3:527–533.
Duan H, Wang Y, Aviram M, et al. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol 1999; 19:3145–3155.
Swaroop M, Wang Y, Miller P, et al. Yeast homolog of human SAG/ROC2/Rbx2/Hrt2 is essential for cell growth, but not for germination: Chip profiling implicates its role in cell cycle regulation. Oncogene 2000; 19:2855–2866.
Zheng N, Schulman BA, Song L, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002; 416:703–709.
Cardozo T, Pagano M . The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 2004; 5:739–751.
Wu K, Chen A, Pan ZQ . Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1-CUL1 complex to promote ubiquitin polymerization. J Biol Chem 2000; 275:32317–32324.
Yen HC, Elledge SJ . Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science 2008; 322:923–929.
Nakayama KI, Nakayama K . Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 2006; 6:369–381.
Silverman JS, Skaar JR, Pagano M . SCF ubiquitin ligases in the maintenance of genome stability. Trends Biochem Sci 2012; 37:66–73.
Jia L, Sun Y . SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets 2011; 11:347–356.
Zhang H, Kobayashi R, Galaktionov K, Beach D . p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell 1995; 82:915–925.
Galan JM, Wiederkehr A, Seol JH, et al. Skp1p and the F-box protein Rcy1p form a non-SCF complex involved in recycling of the SNARE Snc1p in yeast. Mol Cell Biol 2001; 21:3105–3117.
Jourdain I, Spielewoy N, Thompson J, et al. Identification of a conserved F-box protein 6 interactor essential for endocytosis and cytokinesis in fission yeast. Biochem J 2009; 420:169–177.
Piva R, Liu J, Chiarle R, et al. In vivo interference with Skp1 function leads to genetic instability and neoplastic transformation. Mol Cell Biol 2002; 22:8375–8387.
Mandel SA, Fishman-Jacob T, Youdim MB . Modeling sporadic Parkinson's disease by silencing the ubiquitin E3 ligase component, SKP1A. Parkinsonism Relat Disord 2009; 15 Suppl 3:S148–S151.
Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM . cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell 1996; 85:829–839.
Mathias N, Johnson SL, Winey M, et al. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S-phase transition and identifies a conserved family of proteins. Mol Cell Biol 1996; 16:6634–6643.
Duda DM, Borg LA, Scott DC, et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 2008; 134:995–1006.
Goldenberg SJ, Cascio TC, Shumway SD, et al. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell 2004; 119:517–528.
Dealy MJ, Nguyen KV, Lo J, et al. Loss of Cul1 results in early embryonic lethality and dysregulation of cyclin E. Nat Genet 1999; 23:245–248.
Wang Y, Penfold S, Tang X, et al. Deletion of the Cul1 gene in mice causes arrest in early embryogenesis and accumulation of cyclin E. Curr Biol 1999; 9:1191–1194.
Salon C, Brambilla E, Brambilla C, et al. Altered pattern of Cul-1 protein expression and neddylation in human lung tumours: relationships with CAND1 and cyclin E protein levels. J Pathol 2007; 213:303–310.
Chen G, Cheng Y, Martinka M, Li G . Cul1 expression is increased in early stages of human melanoma. Pigment Cell Melanoma Res 2010; 23:572–574.
Singer JD, Gurian-West M, Clurman B, Roberts JM . Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev 1999; 13:2375–2387.
McEvoy JD, Kossatz U, Malek N, Singer JD . Constitutive turnover of cyclin E by Cul3 maintains quiescence. Mol Cell Biol 2007; 27:3651–3666.
Kossatz U, Breuhahn K, Wolf B, et al. The cyclin E regulator cullin 3 prevents mouse hepatic progenitor cells from becoming tumor-initiating cells. J Clin Invest 2010; 120:3820–3833.
Mathew R, Seiler MP, Scanlon ST, et al. BTB-ZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature 2012; 491:618–621.
Siu KT, Rosner MR, Minella AC . An integrated view of cyclin E function and regulation. Cell Cycle 2012; 11:57–64.
Taguchi K, Motohashi H, Yamamoto M . Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011; 16:123–140.
Loignon M, Miao W, Hu L, et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol Cancer Ther 2009; 8:2432–2440.
Yuan WC, Lee YR, Huang SF, et al. A Cullin3-KLHL20 ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell 2011; 20:214–228.
Haagenson KK, Tait L, Wang J, et al. Cullin-3 protein expression levels correlate with breast cancer progression. Cancer Biol Ther 2012; 13:1042–1046.
Hammerman PS, Hayes DN, Wilkerson MD, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489:519–525.
Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012; 482:98–102.
Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292:464–468.
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292:468–472.
Laszlo GS, Cooper JA . Restriction of Src activity by Cullin-5. Curr Biol 2009; 19:157–162.
Lee J, Zhou P . Cullins and cancer. Genes Cancer 2010; 1:690–699.
Sun Y, Li H . Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell 2013; 4:103–116.
Park SW, Chung NG, Hur SY, et al. Mutational analysis of hypoxia-related genes HIF1alpha and CUL2 in common human cancers. APMIS 2009; 117:880–885.
Jackson S, Xiong Y . CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci 2009; 34:562–570.
Liu L, Lee S, Zhang J, et al. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol Cell 2009; 34:451–460.
Li B, Ruiz JC, Chun KT . CUL-4A is critical for early embryonic development. Mol Cell Biol 2002; 22:4997–5005.
Kopanja D, Stoyanova T, Okur MN, et al. Proliferation defects and genome instability in cells lacking Cul4A. Oncogene 2009; 28:2456–2465.
Kopanja D, Roy N, Stoyanova T, et al. Cul4A is essential for spermatogenesis and male fertility. Dev Biol 2011; 352:278–287.
Yin Y, Lin C, Kim ST, et al. The E3 ubiquitin ligase Cullin 4A regulates meiotic progression in mouse spermatogenesis. Dev Biol 2011; 356:51–62.
Jiang B, Zhao W, Yuan J, et al. Lack of Cul4b, an E3 ubiquitin ligase component, leads to embryonic lethality and abnormal placental development. PLoS One 2012; 7:e37070.
Liu L, Yin Y, Li Y, et al. Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Res 2012; 22: 1258–1269.
Wei D, Sun Y . Small RING finger proteins RBX1 and RBX2 of SCF E3 ubiquitin ligases: the role in cancer and as cancer targets. Genes Cancer 2010; 1:700–707.
Schindl M, Gnant M, Schoppmann SF, Horvat R, Birner P . Overexpression of the human homologue for Caenorhabditis elegans cul-4 gene is associated with poor outcome in node-negative breast cancer. Anticancer Res 2007; 27:949–952.
Tarpey PS, Raymond FL, O'Meara S, et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet 2007; 80:345–352.
Zou Y, Liu Q, Chen B, et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet 2007; 80:561–566.
Zhao Y, Sun Y . CUL4B ubiquitin ligase in mouse development: a model for human X-linked mental retardation syndrome? Cell Res 2012; 22:1224–1226.
Li T, Hung MS, Wang Y, et al. Transgenic mice for cre-inducible overexpression of the Cul4A gene. Genesis 2011; 49:134–141.
Arai T, Kasper JS, Skaar JR, et al. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. Proc Natl Acad Sci USA 2003; 100:9855–9860.
Skaar JR, Arai T, DeCaprio JA . Dimerization of CUL7 and PARC is not required for all CUL7 functions and mouse development. Mol Cell Biol 2005; 25:5579–5589.
Huber C, Dias-Santagata D, Glaser A, et al. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet 2005; 37:1119–1124.
Sasaki K, Okamoto N, Kosaki K, et al. Maternal uniparental isodisomy and heterodisomy on chromosome 6 encompassing a CUL7 gene mutation causing 3M syndrome. Clin Genet 2011; 80:478–483.
Maksimova N, Hara K, Miyashia A, et al. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: new population isolate in Asia. J Med Genet 2007; 44:772–778.
Guo G, Gui Y, Gao S, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet 2012; 44:17–19.
Jin J, Cardozo T, Lovering RC, et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 2004; 18:2573–2580.
Cenciarelli C, Chiaur DS, Guardavaccaro D, et al. Identification of a family of human F-box proteins. Curr Biol 1999; 9:1177–1179.
Winston JT, Koepp DM, Zhu C, Elledge SJ, Harper JW . A family of mammalian F-box proteins. Curr Biol 1999; 9:1180–1182.
Yoshida Y, Chiba T, Tokunaga F, et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002; 418:438–442.
Frescas D, Pagano M . Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 2008; 8:438–449.
Harper JW, Tan MK . Ubiquitin pathway proteomics. Mol Cell Proteomics 2012; 11:1541–1550.
Skaar JR, D'Angiolella V, Pagan JK, Pagano M . Snapshot: F box proteins II. Cell 2009; 137:1358, 1358.e1.
Margottin F, Bour SP, Durand H, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1998; 1:565–574.
Spevak W, Keiper BD, Stratowa C, Castanon MJ . Saccharomyces cerevisiae cdc15 mutants arrested at a late stage in anaphase are rescued by Xenopus cDNAs encoding N-ras or a protein with beta-transducin repeats. Mol Cell Biol 1993; 13:4953–4966.
Jiang J, Struhl G . Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 1998; 391:493–496.
Fuchs SY, Spiegelman VS, Kumar KG . The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene 2004; 23:2028–2036.
Nakayama K, Hatakeyama S, Maruyama S, et al. Impaired degradation of inhibitory subunit of NF-kappa B (I kappa B) and beta-catenin as a result of targeted disruption of the beta-TrCP1 gene. Proc Natl Acad Sci USA 2003; 100:8752–8757.
Baguma-Nibasheka M, Kablar B . Abnormal retinal development in the Btrc null mouse. Dev Dyn 2009; 238:2680–2687.
Guardavaccaro D, Kudo Y, Boulaire J, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell 2003; 4:799–812.
Kanarek N, Horwitz E, Mayan I, et al. Spermatogenesis rescue in a mouse deficient for the ubiquitin ligase SCF(beta)-TrCP by single substrate depletion. Genes Dev 2010; 24:470–477.
Kudo Y, Guardavaccaro D, Santamaria PG, et al. Role of F-box protein betaTrcp1 in mammary gland development and tumorigenesis. Mol Cell Biol 2004; 24:8184–8194.
Belaidouni N, Peuchmaur M, Perret C, et al. Overexpression of human beta TrCP1 deleted of its F box induces tumorigenesis in transgenic mice. Oncogene 2005; 24:2271–2276.
Nakayama K, Nagahama H, Minamishima YA, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 2000; 19:2069–2081.
Kossatz U, Dietrich N, Zender L, et al. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev 2004; 18:2602–2607.
Nakayama K, Nagahama H, Minamishima YA, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6:661–672.
Wang J, Han F, Wu J, et al. The role of Skp2 in hematopoietic stem cell quiescence, pool size, and self-renewal. Blood 2011; 118:5429–5438.
Fotovati A, Abu-Ali S, Nakayama K, Nakayama KI . Impaired ovarian development and reduced fertility in female mice deficient in Skp2. J Anat 2011; 218:668–677.
Wu YJ, Sala-Newby GB, Shu KT, et al. S-phase kinase-associated protein-2 (Skp2) promotes vascular smooth muscle cell proliferation and neointima formation in vivo. J Vasc Surg 2009; 50:1135–1142.
Cooke PS, Holsberger DR, Cimafranca MA, et al. The F box protein S phase kinase-associated protein 2 regulates adipose mass and adipocyte number in vivo. Obesity 2007; 15:1400–1408.
Sakai T, Sakaue H, Nakamura T, et al. Skp2 controls adipocyte proliferation during the development of obesity. J Biol Chem 2007; 282:2038–2046.
Suzuki S, Fukasawa H, Kitagawa K, et al. Renal damage in obstructive nephropathy is decreased in Skp2-deficient mice. Am J Pathol 2007; 171:473–483.
Suzuki S, Fukasawa H, Misaki T, et al. The amelioration of renal damage in Skp2-deficient mice canceled by p27 Kip1 deficiency in Skp2-/- p27-/- mice. PLoS One 2012; 7:e36249.
Lin HK, Chen Z, Wang G, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010; 464:374–379.
Agarwal A, Bumm TG, Corbin AS, et al. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood 2008; 112:1960–1970.
Wang H, Bauzon F, Ji P, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/- mice. Nat Genet 2010; 42:83–88.
Old JB, Kratzat S, Hoellein A, et al. Skp2 directs Myc-mediated suppression of p27Kip1 yet has modest effects on Myc-driven lymphomagenesis. Mol Cancer Res 2010; 8:353–362.
Sistrunk C, Macias E, Nakayama K, Kim Y, Rodriguez-Puebla ML . Skp2 is necessary for Myc-induced keratinocyte proliferation but dispensable for Myc oncogenic activity in the oral epithelium. Am J Pathol 2011; 178:2470–2477.
Latres E, Chiarle R, Schulman BA, et al. Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci USA 2001; 98:2515–2520.
Shim EH, Johnson L, Noh HL, et al. Expression of the F-Box protein SKP2 induces hyperplasia, dysplasia, and low-grade carcinoma in the mouse prostate. Cancer Res 2003; 63:1583–1588.
Hartwell LH, Mortimer RK, Culotti J, Culotti M . Genetic control of the cell division cycle in yeast: V. genetic analysis of cdc mutants. Genetics 1973; 74:267–286.
Welcker M, Clurman BE . FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8:83–93.
Tan M, Zhao Y, Kim SJ, et al. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev Cell 2011; 21:1062–1076.
Mao JH, Perez-Losada J, Wu D, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004; 432:775–779.
Tetzlaff MT, Yu W, Li M, et al. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci USA 2004; 101:3338–3345.
Tsunematsu R, Nakayama K, Oike Y, et al. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem 2004; 279:9417–9423.
Onoyama I, Tsunematsu R, Matsumoto A, et al. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med 2007; 204:2875–2888.
Thompson BJ, Jankovic V, Gao J, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med 2008; 205:1395–1408.
Matsuoka S, Oike Y, Onoyama I, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev 2008; 22:986–991.
Inuzuka H, Shaik S, Onoyama I, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011; 471:104–109.
Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011; 471:110–114.
Sancho R, Jandke A, Davis H, et al. F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology 2010; 139:929–941.
Onoyama I, Suzuki A, Matsumoto A, et al. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. J Clin Invest 2011; 121:342–354.
Hoeck JD, Jandke A, Blake SM, et al. Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci 2010; 13:1365–1372.
Matsumoto A, Onoyama I, Sunabori T, et al. Fbxw7-dependent degradation of Notch is required for control of “stemness” and neuronal-glial differentiation in neural stem cells. J Biol Chem 2011; 286:13754–13764.
Wang Z, Inuzuka H, Zhong J, et al. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett 2012; 586:1409–1418.
Xu X, Sarikas A, Dias-Santagata DC, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell 2008; 30:403–414.
Okabe H, Lee SH, Phuchareon J, et al. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS One 2006; 1:e128.
Tsunematsu R, Nishiyama M, Kotoshiba S, et al. Fbxw8 is essential for Cul1-Cul7 complex formation and for placental development. Mol Cell Biol 2006; 26:6157–6169.
Tsutsumi T, Kuwabara H, Arai T, Xiao Y, Decaprio JA . Disruption of the Fbxw8 gene results in pre- and postnatal growth retardation in mice. Mol Cell Biol 2008; 28:743–751.
Lin DI, Barbash O, Kumar KG, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell 2006; 24:355–366.
Barbash O, Zamfirova P, Lin DI, et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell 2008; 14:68–78.
Pontano LL, Aggarwal P, Barbash O, et al. Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability. Mol Cell Biol 2008; 28:7245–7258.
Vaites LP, Lee EK, Lian Z, et al. The Fbx4 tumor suppressor regulates cyclin D1 accumulation and prevents neoplastic transformation. Mol Cell Biol 2011; 31:4513–4523.
Gong B, Chen F, Pan Y, et al. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer's disease amyloidosis and improves synaptic function. Aging Cell 2010; 9:1018–1031.
Nelson RF, Glenn KA, Zhang Y, et al. Selective cochlear degeneration in mice lacking the F-box protein, Fbx2, a glycoprotein-specific ubiquitin ligase subunit. J Neurosci 2007; 27:5163–5171.
Hsu JM, Lee YC, Yu CT, Huang CY . Fbx7 functions in the SCF complex regulating Cdk1-cyclin B-phosphorylated hepatoma up-regulated protein (HURP) proteolysis by a proline-rich region. J Biol Chem 2004; 279:32592–32602.
Meziane el K, Randle SJ, Nelson DE, Lomonosov M, Laman H . Knockdown of Fbxo7 reveals its regulatory role in proliferation and differentiation of haematopoietic precursor cells. J Cell Sci 2011; 124:2175–2186.
Vashisht AA, Zumbrennen KB, Huang X, et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009; 326:718–721.
Moroishi T, Nishiyama M, Takeda Y, Iwai K, Nakayama KI . The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab 2011; 14:339–351.
Saiga T, Fukuda T, Matsumoto M, et al. Fbxo45 forms a novel ubiquitin ligase complex and is required for neuronal development. Mol Cell Biol 2009; 29:3529–3543.
Peschiaroli A, Scialpi F, Bernassola F, Pagano M, Melino G . The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009; 28:3157–3166.
Tokuzawa Y, Kaiho E, Maruyama M, et al. Fbx15 is a novel target of Oct3/4 but is dispensable for embryonic stem cell self-renewal and mouse development. Mol Cell Biol 2003; 23:2699–2708.
Swaroop M, Bian J, Aviram M, et al. Expression, purification, and biochemical characterization of SAG, a RING finger redox sensitive protein. Free Radic Biol Med 1999; 27:193–202.
Swaroop M, Gosink M, Sun Y . SAG/ROC2/Rbx2/Hrt2, a component of SCF E3 ubiquitin ligase: genomic structure, a splicing variant, and two family pseudogenes. DNA Cell Biol 2001; 20:425–434.
Chen A, Wu K, Fuchs SY, et al. The conserved RING-H2 finger of ROC1 is required for ubiquitin ligation. J Biol Chem 2000; 275:15432–15439.
Ohta T, Michel JJ, Xiong Y . Association with cullin partners protects ROC proteins from proteasome-dependent degradation. Oncogene 1999; 18:6758–6766.
Gu Q, Tan M, Sun Y . SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradecanoylphorbol-13-acetate-induced neoplastic transformation. Cancer Res 2007; 67:3616–3625.
Kamura T, Maenaka K, Kotoshiba S, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev 2004; 18:3055–3065.
Tan M, Davis SW, Saunders TL, Zhu Y, Sun Y . RBX1/ROC1 disruption results in early embryonic lethality due to proliferation failure, partially rescued by simultaneous loss of p27. Proc Natl Acad Sci USA 2009; 106:6203–6208.
Morimoto M, Nishida T, Nagayama Y, Yasuda H . Nedd8-modification of Cul1 is promoted by Roc1 as a Nedd8-E3 ligase and regulates its stability. Biochem Biophys Res Commun 2003; 301:392–398.
Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K . Nedd8 on cullin: building an expressway to protein destruction. Oncogene 2004; 23:1985–1997.
Tan M, Zhu Y, Kovacev J, et al. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-κB activation in mouse embryonic stem cells. Free Radic Biol Med 2010; 49:976–983.
Kohroki J, Nishiyama T, Nakamura T, Masuho Y . ASB proteins interact with Cullin5 and Rbx2 to form E3 ubiquitin ligase complexes. FEBS Lett 2005; 579:6796–6802.
Sun Y . Induction of glutathione synthetase by 1,10-phenanthroline. FEBS Lett 1997; 408:16–20.
Tan M, Gu Q, He H, et al. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1alpha ubiquitination and degradation. Oncogene 2008; 27:1404–1411.
Gu Q, Bowden GT, Normolle D, Sun Y . SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol 2007; 178:1009–1023.
He H, Gu Q, Zheng M, Normolle D, Sun Y . SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis 2008; 29:858–865.
Kemp CJ . Multistep skin cancer in mice as a model to study the evolution of cancer cells. Semin Cancer Biol 2005; 15:460–473.
Cichowski K, Jacks T . NF1 tumor suppressor gene function: narrowing the GAP. Cell 2001; 104:593–604.
Jia L, Yang J, Hao X, et al. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clin Cancer Res 2010; 16:814–824.
Huang Y, Duan H, Sun Y . Elevated expression of SAG/ROC2/Rbx2/Hrt2 in human colon carcinomas: SAG does not induce neoplastic transformation, but its antisense transfection inhibits tumor cell growth. Mol Carcinog 2001; 30:62–70.
Sasaki H, Yukiue H, Kobayashi Y, et al. Expression of the sensitive to apoptosis gene, SAG, as a prognostic marker in non small cell lung cancer. Int J Cancer 2001; 95:375–377.
Jia L, Soengas MS, Sun Y . ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res 2009; 69:4974–4982.
Yang D, Li L, Liu H, et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ 2012; 20:235–247.
Sun Y . Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol Ther 2003; 2:623–629.
Sun Y . E3 ubiquitin ligases as cancer targets and biomarkers. Neoplasia 2006; 8:645–654.
Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 2010; 37:102–111.
Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009; 458:732–736.
Milhollen MA, Traore T, Adams-Duffy J, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-(kappa)B-dependent lymphoma. Blood 2010; 116:1515–1523.
Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood 2010; 115:3796–3800.
Tan M, Li Y, Yang R, Xi N, Sun Y . Inactivation of SAG E3 ubiquitin ligase blocks embryonic stem cell differentiation and sensitizes leukemia cells to retinoid acid. PLoS One 2011; 6:e27726.
Milhollen MA, Narayanan U, Soucy TA, et al. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res 2011; 71:3042–3051.
Jia L, Li H, Sun Y . Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia 2011; 13:561–569.
Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A . NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res 2010; 70:10310–10320.
Luo Z, Yu G, Lee HW, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res 2012; 72:3360–3371.
Zhao Y, Xiong X, Jia L, Sun Y . Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis 2012; 3:e386.
Nawrocki ST, Griffin P, Kelly KR, Carew JS . MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs 2012; 21:1563–1573.
Di Fonzo A, Dekker MC, Montagna P, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009; 72:240–245.
Deng H, Liang H, Jankovic J . F-Box only protein 7 gene in Parkinsonian-Pyramidal disease. Arch Neurol 2012; 1:1–5.
Jia L, Bickel JS, Wu J, et al. RBX1 (RING-box protein 1) E3 ubiquitin ligase is required for genomic integrity by modulating DNA replication licensing proteins. J Biol Chem 2011; 286:3379–3386.
Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE . The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 2010; 1:708–716.
Acknowledgements
This work was supported by the NCI grants (CA118762, CA156744, CA170995, and CA171277) to YS and the NIGMS grant (GM094777) to WW. We apologize to the authors whose original studies were not cited due to the space limitation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article
Cite this article
Zhou, W., Wei, W. & Sun, Y. Genetically engineered mouse models for functional studies of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases. Cell Res 23, 599–619 (2013). https://doi.org/10.1038/cr.2013.44
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.44
Keywords
This article is cited by
-
The phosphatase DUSP22 inhibits UBR2-mediated K63-ubiquitination and activation of Lck downstream of TCR signalling
Nature Communications (2024)
-
Genome-wide analysis of genes encoding core components of the ubiquitin system during cerebral cortex development
Molecular Brain (2022)
-
Functional characterization of FBXL7 as a novel player in human cancers
Cell Death Discovery (2022)
-
The Ube2m-Rbx1 neddylation-Cullin-RING-Ligase proteins are essential for the maintenance of Regulatory T cell fitness
Nature Communications (2022)
-
Pseudophosphatase STYX is induced by Helicobacter pylori and promotes gastric cancer progression by inhibiting FBXO31 function
Cell Death & Disease (2022)
