
Abstract
TRIB2, a serine/threonine pseudokinase identified as an oncogene, is expressed at high levels in the T-cell compartment of hematopoiesis. The proliferation of developing thymocytes is tightly controlled to prevent leukemic transformation of T cells. Here we examine Trib2 loss in murine hematopoiesis under steady state and proliferative stress conditions, including genotoxic and oncogenic stress. Trib2−/− developing thymocytes show increased proliferation, and Trib2−/− mice have significantly higher thymic cellularity at steady state. During stress hematopoiesis, Trib2−/− developing thymocytes undergo accelerated proliferation and demonstrate hypersensitivity to 5-fluorouracil (5-FU)-induced cell death. Despite the increased cell death post 5-FU-induced proliferative stress, Trib2−/− mice exhibit accelerated thymopoietic recovery post treatment due to increased cell division kinetics of developing thymocytes. The increased proliferation in Trib2−/− thymocytes was exacerbated under oncogenic stress. In an experimental murine T-cell acute lymphoblastic leukemia (T-ALL) model, Trib2−/− mice had reduced latency in vivo, which associated with impaired MAP kinase (MAPK) activation. High and low expression levels of Trib2 correlate with immature and mature subtypes of human T-ALL, respectively, and associate with MAPK. Thus, TRIB2 emerges as a novel regulator of thymocyte cellular proliferation, important for the thymopoietic response to genotoxic and oncogenic stress, and possessing tumor suppressor function.
Similar content being viewed by others

Introduction
TRIB2 is a member of the mammalian Tribbles family of serine/threonine pseudokinases (TRIB1–3). Studies focused on the pathological role of TRIB2 in various disease states, including hematological malignancy, solid tumors, autoimmune and inflammatory diseases, have identified TRIB2 as a critical signaling modulator and mediator [1]. However, it is unclear if this is true in a physiological context where regulation of diverse signaling pathways is cell type and developmental stage dependent. Studies of Tribbles orthologues in Xenopus [2] and Drosophila [3–5] highlight an evolutionary conserved role for Tribbles in the regulation of normal cellular proliferation. In these organisms, Tribbles coordinates cell division and morphogenesis to ensure proper organ development. In Drosophila, Tribbles ensuring mitosis occurs in a timely manner by regulating String/CDC25 turnover at protein level [3–5]. Histological study of a Trib2 knockout mouse model showed no phenotypic defects in various organs during embryogenesis [6]. These data suggest TRIB2 function is required for proper cellular behavior rather than tissue organization.
In hematological malignancies, TRIB2 has been implicated in acute myeloid leukemia (AML), as well as T-cell acute lymphoblastic leukemia (T-ALL). Enforced expression of Trib2 by retroviral transduction induces potent murine AML [7] and Trib2 is a target gene of MEIS1 [8] in HOX-induced murine leukemia. In human AML, elevated expression of Trib2 was shown to be driven by the transcription factors E2F1 [9] and NOTCH1 [10] where the latter was found to be aberrantly activated in an AML subset that has a mixed myeloid/T-lymphoid phenotype [10]. In human T-ALL, high Trib2 expression was found to be associated with activated NOTCH1 signaling [11]. Indeed, Trib2 was also identified as a downstream target of PITX1 [12] and TAL1 [13], transcription factors that are also aberrantly expressed in T-ALL. These oncogenic transcription factors involved in TRIB2 regulation and associated AML and T-ALL are important players in lineage specification during normal hematopoiesis [14–18], suggesting a potential role for TRIB2 in normal hematopoiesis.
We previously showed that Trib2 expression is highest in the T-cell lineage in normal hematopoiesis and is regulated during αβ T-cell development in the thymus [19]. The thymus is a lymphoid organ where progenitors from the bone marrow (BM) commit to T-cell lineage development, differentiate into functional naive T cells, and are then exported to the periphery as part of the adaptive immune system. Here we show that TRIB2 is a novel cell cycle brake essential for balanced proliferation of developing murine thymocytes. Loss of TRIB2 causes developing thymocytes to be highly proliferative but with heightened sensitivity to in vivo genotoxic drug treatment. As such, Trib2 ablation accelerates thymopoietic recovery following genotoxic insult. However, in cells expressing a T-cell oncogene, the absence of TRIB2 led to enhanced T-cell leukemic transformation associated with impaired MAP kinase (MAPK) signaling. In the human disease, low levels of Trib2 expression correlate with a mature T-ALL phenotype and high levels of expression with an immature phenotype. In accordance with our experimental model, MAPK signaling correlates with Trib2 expression in T-ALL.
Results
TRIB2 is dispensable for murine hematopoiesis in the bone marrow
To investigate whether TRIB2 has a role in normal hematopoiesis, we examined the hematopoietic system of a Trib2 knockout mouse model (129S5-Trib2tm1Lex, referred as Trib2−/− hereafter) where the coding and noncoding regions of exon 1 were disrupted and thus enabled Trib2 genotyping by PCR analysis (Supplementary Figure S1; Supplementary Table S1). Compared with wild-type (WT) mice, Trib2−/− mice had similar red blood cell and white blood cell differential counts, except for the platelet count which was significantly higher but within the normal physiological range (Figure 1a and b). In addition, no difference was found between WT and Trib2−/− mice in the distribution of mature myeloid, B and T-cells in the blood (Figure 1c). These data indicate that loss of TRIB2 does not affect terminal differentiation and production of mature blood cells of different lineages. We next studied the BM, the primary site of adult hematopoiesis, to determine whether TRIB2 influences hematopoietic cell fate choice at an earlier stage. Trib2−/− mice had similar BM cellularity compared with WT mice (Figure 1d). No difference was found in the frequency of circulating CD4+ and CD8+ T-cells, lineage committed Gr-1+/CD11b+ myeloid cells and CD19+/B220+ B-lymphoid cells (Figure 1e). Moving up the hematopoietic hierarchy, Trib2−/− mice had similar frequencies of hematopoietic stem and progenitor cells (HSPCs), including multipotent progenitors, common lymphoid progenitors, granulocyte-macrophage progenitors, megakaryocyte-erythroid progenitors, common lymphoid progenitors and hematopoietic stem cells (HSCs) compared with WT mice (Figure 1f and g). Hence, TRIB2 is not essential for hematopoietic cell fate choice as loss of TRIB2 did not lead to skewing of hematopoietic cell differentiation. Thus, at steady state, Trib2 ablation does not affect the hierarchical organization of the murine hematopoietic system.

TRIB2 loss does not affect murine hematopoiesis in bone marrow. Complete blood counts (a) and WBC differential counts (b) of WT (n=13) and Trib2−/− (n=22) mice were determined by hematology analyzer. BA, basophils; EO, eosinophils; LY, lymphocytes; MO, monocytes; NE, neutrophils. (c) The distribution of mature myeloid, B and T-cells in the blood of WT (n=3) and Trib2−/− (n=10) mice were measured by flow cytometry using the indicated lineage specific cell surface markers. (d) BM cellularity (n=5–7 per genotype) was counted by trypan blue exclusion after RBC lysis of the cell suspension collected from two pelvises, femurs and tibias. (e) The distribution of myeloid, B and circulating T-cells in the BM of WT (n=3) and Trib2−/− (n=9) mice. (f) Immunophenotyping of HSPCs populations (HSC, MPP, CMP, GMP, MEP and CLP) in BM. Each sub-population is indicated in the outlined areas (top row). The corresponding values in the representative staining profile of WT (middle row) and Trib2−/− (bottom row) mice (n=3 per genotype) are frequency of BM and graphed in g. CLP, Lin−IL-7Rα+; CMP, LK CD34+CD16/32lo; GMP, LK CD34+CD16/32hi; HSC, LSK CD150+CD48−; Lin, lineage; LK, Lin−c-Kit+; LSK, Lin−Sca-1+c-Kit+; MPP, LSK CD150−CD48−; MEP, LK CD34−CD16/32−; SSC-H, side scatter-height. For a, unpaired Student’s t-test was used for statistical analysis. *P<0.05, all quantified data are presented as mean and s.e.m. CMP, common myeloid progenitor; CLP, common lymphoid progenitor; GMP, granulocyte-macrophage progenitor; MEP, megakaryocyte-erythroid progenitor; MPP, multipotent progenitor; RBC, red blood cell; WBC, white blood cell.
We next assessed the repopulating capability and multi-lineage differentiation potential of Trib2−/− HSPCs by transplantation of CD45.2+ whole-BM-nucleated cells (WT or Trib2−/− donor) into lethally irradiated CD45.1+ mice (recipients). TRIB2 loss did not affect long-term engraftment of donor cells and their capability to fully reconstitute the blood system of recipients (Supplementary Figure S2A–C). Analysis of blood collected periodically from recipients showed no skewing of differentiation into myeloid and lymphoid lineages in the absence of TRIB2 during hematopoietic reconstitution (Supplementary Figure S2D). No significant difference was found in the HSPC populations in recipients transplanted with either genotype (Supplementary Figure S2E and F). We conclude that TRIB2 is dispensable for the maintenance of the hematopoietic system through differentiating HSPC populations that reside in the BM.
TRIB2 regulates the proliferation of developing thymocytes
T-cell development takes place in the thymus that is seeded constantly by T-cell progenitors originating in the BM. We found that Trib2−/− mice had statistically significant higher thymic cellularity compared with WT mice (Figure 2a). This suggests dysregulation of thymopoiesis in the absence of TRIB2. We further examined thymic subsets along the αβ T-lineage developmental pathway by immunophenotyping. Non-T-lineage markers (CD11c, Gr-1, B220, Ter119 and NK1.1) were included in all the experimental analysis to exclude cells of other lineages present in thymus. Gating for lineage markers, including CD4 and CD8, were adjusted so as not to exclude c-Kit+ thymic progenitors that express low cell surface levels of lineage markers when defining immature CD4−CD8− double-negative (DN) thymocytes [20] (Supplementary Figure S3). For gating of thymic subsets, DN3 thymocytes (LinloCD44−CD25+) were divided further into DN3E (expected: FSClo) and DN3L (larger: FSChi) subsets, based on cell size [21]. This is equivalent to characterization of DN3 into DN3a and DN3b subsets, based on CD27 marker, that corresponded to pre- and post-β selection [22]. Similarly, CD4+CD8+ double-positive (DP) thymocytes were divided further into DPsm (small resting: FSClo) and DPbl (blasts: FSChi) subsets [23]. As described [21, 24, 25], we found that DN3E and DPsm subsets were not proliferative as >99% were in G0/G1 phase and only 10–20% were CD71+. In contrast, DN3L and DPbl subsets were actively cycling as 60–70% were in S–G2/M phases and 90% were CD71+ (Supplementary Figure S4A–C). Our analysis of the thymic subsets showed that, in general, Trib2−/− mice had a lower frequency of immature DN1–4 subsets, but the mature DP and single-positive subsets were unaffected compared with WT mice (Figure 2b and c). The higher cellularity of Trib2−/− thymus was due to the significant increase of mature subsets (DPsm and CD4 single positive) but not the immature DN1–4 subsets (Figure 2d). The DN1 (LinloCD44+CD25−) subset is heterogeneous and divided into DN1a–e subsets based on CD24 and c-Kit surface expression [26]. Here the DN1 subset was divided broadly into c-Kithi (DN1a/b), c-Kitlo (DN1c) and c-Kit− (DN1d/e) subsets (Figure 2e). Trib2−/− c-Kit− DN1 progenitors were present at a significantly lower frequency but had similar cell numbers compared with WT mice (Figure 2f and g). To rule out the possibility that lower frequency of Trib2−/− DN1–4 subsets were due to increased apoptosis, we measured Annexin V expression and showed no difference between the two genotypes (Figure 2h). As Trib2−/− mice had higher numbers of mature subsets that must be derived from immature subsets, we hypothesized that immature thymocytes were cycling faster and gave rise to more mature differentiated thymocytes in the absence of TRIB2. Ki-67 is a proliferation marker, expressed by cells in active cell cycle (G1–S–G2/M) but not by resting cells in G0 [27]. Indeed, intracellular staining of Ki-67 showed that Trib2−/− DN1, DN2 and DN3E subsets had significantly higher levels of Ki-67 compared with WT thymic subsets (Figure 2i), indicating that more Trib2−/− developing thymocytes are in cycling state. This does not result in T-cell accumulation outside the thymus however (Supplementary Figure S5).

TRIB2 regulates the homeostasis of intrathymic T-cell development. (a) Thymic cellularity of WT (n=31) and Trib2−/− (n=21) mice was counted by trypan blue exclusion after RBC lysis. (b) Flow cytometry of thymic subsets. The complete gating strategy is provided in Supplementary Figure S3. Each subset is indicated in the outlined areas (top row). The corresponded values in the representative staining profile of WT (middle row) and Trib2−/− (bottom row) mice are frequency of thymus and graphed in c. CD4 SP, CD4+CD8−; CD8 SP, CD4−CD8+; DN1, LinloCD44+CD25−; DN2, LinloCD44+CD25+; DN3E, LinloCD44−CD25+FSClo; DN3L, LinloCD44−CD25+FSChi; DN4, LinloCD44−CD25−; DPbl, CD4+CD8+FSChi; DPsm, CD4+CD8+FSClo; FSC-A, forward scatter-area; SSC-A, side scatter-area. (d) Number of cells for each subset. (e) Further characterization of DN1 cells based on c-Kit surface expression. hi, high; lo, low; Neg, negative. DN1 subsets were graphed in frequency of thymus (f) and cell number (g). For c, d, f and g, n=7–8 per genotype. (h) Basal level of apoptosis of each thymic subset (n=3 per genotype) was determined by the surface expression of Annexin V after exclusion of DAPI-stained dead cells. (i) Intracellular level of Ki-67 across thymic subsets (n=4–5 per genotype) was measured by flow cytometry (left). An overlap of histogram (right) showed Trib2−/− DN1 cells had higher level of Ki-67 compared with that of WT. FMO, Fluorescence Minus One; Norm, normalized. For statistical analyses, unpaired Student’s t-test was used for a, i, and two-way ANOVA was used for c, d and f. **P<0.01; ***P<0.001, all quantified data are presented as mean and s.e.m. ANOVA, analysis of variance; DAPI, 4′, 6-diamidino-2-phenylindole; RBC, red blood cell; SP, single positive.
Cell cycle regulation is crucial for proper T-cell development. DN3 thymocytes must be briefly arrested (DN3E) at the G0/G1 cell cycle phases to allow V(D)J recombination to take place at the Tcrb locus, initiated by the RAG proteins [28, 29]. Functional Tcrb rearrangements lead to formation of pre-T-cell receptors where the signals drive the subsequent development of thymocytes to early DP stage. Given that Trib2−/− developing thymocytes proliferate faster, we determined whether the loss of TRIB2 affects the cell cycle status of these subsets and Tcrb rearrangements. DNA staining showed that Trib2−/− thymocytes at each stage of the T-cell development had a similar cell cycle profile compared with WT mice (Supplementary Figure S4A and B). This suggests that TRIB2 does not regulate the cell cycle phase progression, and that the increased cycling of Trib2−/− developing thymocytes is likely due to changes in cell division kinetics at the steady state. Importantly, Trib2−/− DN3E thymocytes were arrested at G0/G1 phases such as WT DN3E thymocytes. As such, analysis of Tcrb rearrangement involving the Jβ2.1 to Jβ2.7 gene segments demonstrated that Trib2−/− thymocytes contain polyclonal Tcrb rearrangements similar to WT thymocytes (Supplementary Figure S4D).
Trib2−/− thymocytes are hypersensitive to 5-fluorouracil-induced cell death
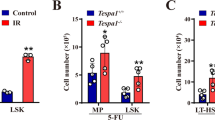
5-fluorouracil (5-FU) is a standard chemotherapeutic drug widely used to treat cancers, as it targets rapidly cycling cells. Misincorporation of 5-FU metabolite into DNA during DNA replication initiates futile cycles of DNA excision, repair and further misincorporation that eventually lead to DNA strand breaks and cell death [30]. On account of that, we hypothesized that Trib2−/− developing thymocytes which are highly proliferative to be more sensitive to 5-FU compared with WT thymocytes [31]. We performed a time-course study of in vivo 5-FU treatment. Trib2−/− mice had significantly higher total thymic cellularity at steady state (Figure 2a); however, the significance difference lost at 16 and 24 h post treatment (Figure 3a). Significant increase of cell death (late apoptotic) was found in Trib2−/− thymic cells at 24 h post treatment (Figure 3b and c). Notably, we demonstrated that DN3L and DPbl subsets, which are known to be proliferative (Supplementary Figure S4A–C), were significantly reduced in Trib2−/− mice 16 h post treatment. DN3E and DPsm subsets, which were in resting state, were unaffected in treated WT and Trib2−/− mice. Unexpectedly, Trib2−/− c-Kithi DN1 thymic progenitors were also significantly reduced after 16 h of exposure to 5-FU, whereas Trib2−/− c-Kit− and c-Kitlo DN1 progenitors were unaffected like WT mice (Figure 3d and e). c-Kithi DN1 progenitors were shown previously to exclusively exhibit a proliferative burst capacity by OP9-DL1 co-culture system compared with c-Kit− and c-Kitlo DN1 progenitors [26]. Interestingly, 5-FU was shown recently to preferentially induce apoptosis in c-Kithi HSCs that have rapid cell division kinetics compared with c-Kitlo HSCs in vitro [32]. Overall, these data confirmed a role for TRIB2 in controlling the cell division kinetics of thymocytes and hence their enhanced sensitivity to 5-FU genotoxic drug.

Trib2−/− thymocytes are more susceptible to 5-FU-induced cell death. (a) Mice were treated with 5-FU (250 mg kg−1, i.p.) and sacrificed after 16 and 24 h to determine the thymic cellularity. (b) Apoptosis assays were performed to determine the fraction of living, early and late apoptotic cells in thymus at 16 and 24 h post treatment. (c) A representative staining profile (b) of 5-FU-treated WT and Trib2−/− thymus. Living, DAPI−AV−; early apoptotic, DAPI−AV+; late apoptotic, DAPI+AV+. The values indicated in the outlined areas are frequency of thymus. (d) The cellularity of thymic subsets at 16 h post treatment. (e) Overlay of the histograms of DN1 and DN3 cells for c-Kit expression and FSC-A signals, respectively, showed loss of Trib2−/− DN1c-Kit+ and DN3L cells at 16 h post treatment. For a, b and d, n=5 per genotype per studied time point. For statistical analyses, unpaired Student’s t-test was used for a and two-way ANOVA was used for b, d. *P<0.05; **P<0.01, all quantified data are presented as mean and s.e.m. ANOVA, analysis of variance; DAPI, 4’, 6-diamidino-2-phenylindole; i.p., intraperitoneal.
Acceleration of thymopoietic recovery in the absence of TRIB2 after genotoxic insult
Previous studies on the response of thymus to 5-FU-mediated genotoxic insult have shown that 5-FU induces apoptosis of thymocytes, thymic weight loss and damages to thymic architecture that is crucial for T-cell development [33, 34]. Thymic inner architecture exhibited morphological recovery on day 7 and was back to normal on day 10 post 5-FU injury in previous work. However, little is known about thymopoietic restoration. We compared thymopoietic recovery of WT and Trib2−/− mice 4 and 14 days after a single dose of 5-FU administration. Trib2−/− mice had higher thymic cellularity compared with WT mice at 4 and 14 days post treatment and the thymus was relatively bigger at 14 days post treatment (Figure 4a and b). Importantly, the higher cell count was not due to the increase of myeloid and B cells that do not normally develop in thymus (Figure 4c). Although WT and Trib2−/− mice both had the hierarchy of thymic subsets restored to normal on day 14 compared with day 4 post 5-FU treatment (Supplementary Figure S6A–C), Trib2−/− mice had significantly higher numbers of DN1 progenitors and the subsequent mature subsets (Figure 4d). This indicates that Trib2−/− mice had accelerated thymopoietic recovery. At steady state, thymopoiesis is sustained mainly by c-Kithi DN1 progenitors [35]. However, these progenitors were absent in WT and Trib2−/− thymus from day 1 to day 14 post 5-FU treatment (Supplementary Figure S6D). Instead, we found a significant increase of c-Kit− DN1 progenitors in Trib2−/− thymus suggesting expansion of these progenitors drives the accelerated recovery (Figure 4e).

Thymopoietic recovery is accelerated in the absence of TRIB2 after genotoxic stress. (a) Thymic cellularity of mice after 4 and 14 days of 5-FU treatment. (b) The white arrow indicates representative thymus of the dissected WT and Trib2−/−mice. (c) The cellularity of non-T-lineage cells (DC, CD11c+; NK cells, Nk1.1+; B, CD19+B220+; Myeloid, Gr-1+CD11b+) in thymus (n=3 per genotype) were determined by flow cytometry. Cellularity for thymic subsets (d) and DN1 subsets (e). (f) PCR analysis (n=3 per genotype) of Tcrb rearrangement involving the Dβ2 to Jβ2 gene segments. GL denotes the position of the germline PCR product and number indicates the different rearrangements. 1 kb M, 1 kb DNA marker; neg ctrl, no template negative control. (g) Flow cytometry to determine the fraction of developing thymocytes in resting (G0) and active (G1–S/G2/M) cell cycle. Each phase is indicated in the outlined areas (top row). The corresponding values in the representative staining profile of WT (middle row) and Trib2−/− (bottom row) mice (n=2–3 per genotype) are frequency of each phase and graphed in h. (i) The frequency of BrdU uptake by DN3 thymocytes after 1 and 4 h of pulsing. A representative staining profile of WT and Trib2−/− mice (n=3 per genotype per studied time point) is shown here. The values indicated in the outlined areas are the frequencies of BrdU+ DN3 thymocytes and graphed in j. For a, d and e, n=9 per genotype per studied time point. 5-FU dosage for a–e was 250 mg kg−1, whereas for f–j it was 200 mg kg−1. For statistical analyses, unpaired Student’s t-test was used for a and two-way ANOVA was used for c–e, h and j. *P<0.05; **P<0.01; ***P<0.001, all quantified data are presented as mean and s.e.m. ANOVA, analysis of variance.
We postulated that the acceleration of thymopoietic recovery in the absence of TRIB2 is due to the intrinsic highly proliferative nature of Trib2−/− thymocytes. In developing thymocytes, proliferation and differentiation are tightly linked. Analysis of Tcrb rearrangements for the joining of Dβ2 to Jβ2 gene segments showed less immature thymocytes that have not undergone Tcrb rearrangements (germline (GL) band) present in Trib2−/− thymus compared with WT thymus 14 days post 5-FU treatment (Figure 4f). This supports our finding that thymopoiesis was more active in treated Trib2−/− mice. We further assessed the quiescent state of DN thymocytes through dual staining of DNA and Ki-67. Compared with WT DN thymocytes, significantly less Trib2−/− thymocytes in DN1 and DN4 subsets were resting (G0) and correspondingly more Trib2−/− thymocytes in these subsets were in cycling state (G1–S/G2–M; Figure 4g and h). This indicates that TRIB2 regulates the cell cycle entry of thymocytes. To expand beyond a static assessment of proliferation, we did time-course experiments of in vivo bromodeoxyuridine (BrdU) pulsing in WT and Trib2−/− mice. DN3 thymocytes of both genotypes had similar uptake of BrdU after 1 h of pulsing; however, BrdU+ Trib2−/− DN3 thymocytes were significantly increased after 4 h of pulsing (Figure 4i and j). This demonstrated that Trib2−/− developing thymocytes had higher cell division kinetics and more DN3 thymocytes were available to uptake BrdU while replicating their DNA.
It is noteworthy that TRIB2 loss only affected thymopoietic recovery but not hematopoietic regeneration in BM. On day 14 post 5-FU treatment, blood cell counts of WT and Trib2−/− mice were restored to normal with no measurable differences detected (Supplementary Figure S7A and B). However, more CD4+ and CD8+ T cells were present in the blood of Trib2−/− mice though these were not statistically significant (Supplementary Figure S7C). Both genotypes had similar BM cellularity and HSPC populations, including common lymphoid progenitors that are lymphoid-restricted progenitors [36] (Supplementary Figure S7D–F). Hence, hematopoiesis in BM was restored normally in the absence of TRIB2.
TRIB2 loss accelerates T-ALL via defective MAPK signaling
As TRIB2 appears to negatively regulate the proliferation of developing thymocytes, the normal counterpart of T-ALL, we examined the role of TRIB2 in T-cell leukemogenesis using a NOTCH1-induced T-ALL BM transplantation mouse model [37]. WT and Trib2−/− BM cells were transduced with an empty vector control (MigR1) or a retroviral vector encoding an intracellular Notch1 transgene (ICN1). The cells, which were CD45.2+, were then transplanted into lethally irradiated CD45.1+ recipient mice to monitor the development of T-ALL. All experimental groups were verified by Trib2 genotyping analysis of the transplanted moribund mice (Supplementary Figure S8). Mice transplanted with ICN1-transduced Trib2−/− donor cells succumbed to T-ALL disease with a shorter latency (median survival of 43.5 days), whereas mice transplanted with ICN1-transduced WT donor cells had a median survival of 63.5 days. Kaplan–Meier survival analysis for these two groups was significantly different (Figure 5a), indicating that T-ALL onset driven by NOTCH1 overexpression was accelerated in the absence of TRIB2. Green fluorescent protein expression was used as a marker for transduced donor cells and analysis showed similar engraftment levels of transduced WT and Trib2−/− donor cells in the recipients from all groups across different analyzed organs (Figure 5b). TRIB2 loss did not alter the phenotype of murine NOTCH1-induced T-ALL (Figure 5c). Despite a difference of 20 days for disease onset, mice that succumbed earlier to Trib2−/− T-ALL had a similar degree of leukemic burden (Figure 5d and e) but exhibited a trend of higher organ infiltration of leukemic cells (Figure 5f) compared with mice that succumbed to WT T-ALL. We have ruled out that the T-ALL phenotype is not due to an increase in Trib1 expression, as Trib1 expression levels do not change upon knockdown of Trib2 in T-ALL or AML cell lines (Supplementary Figure S9; Supplementary Table S2). Tribbles family (TRIB1–3) is known to be associated with MAPK signaling and required for the activation of ERK, JNK and p38 [38–41]. Western blotting for MAPK signals in leukemic infiltrated BM samples showed impaired activation of ERK, JNK and p38 in Trib2−/− T-ALL compared with WT T-ALL (Figure 5g).

TRIB2 attenuates the leukemia initiation potential of NOTCH1. (a) Kaplan–Meier survival analysis of the lethally irradiated recipient (CD45.1+) mice transplanted with 5-FU-enriched WT or Trib2−/− BM cells (donor: CD45.2+) transduced with either MigR1 control or ICN1 transgene. The number (n) of mice analyzed was from three independent experiments. Engraftment of transduced donor cells (CD45.2+GFP+) (b) and development of T-ALL leukemia (CD4+CD8+) (c) in moribund mice were verified by flow cytometry analysis. The values in the outlined areas (c: bottom row) are frequency of leukemic cells in the BM of moribund mice. Leukemic burden was assessed by the spleen to body weight ratio (d) and WBC counts (e) of moribund mice. (f) Leukemic infiltration was assessed by measurement of the frequency of leukemic cells in various organs of moribund mice. (g) Activation of MAPK signaling in T-ALL was determined by western blotting for p-Erk, total Erk, p-p38, total p38, p-JNK, JNK1 and β-actin signals in the leukemic BM (n=4 per group). Signals shown were developed from triplicate immunoblots. For a, Log-rank test was used to compare the survival curves. **P=0.0053, all quantified data are presented as mean and s.e.m. GFP, green fluorescent protein; WBC, white blood cell.
Trib2 expression levels distinguish molecular subtypes of T-ALL and correlate with MAPK signaling
As TRIB2 loss accelerated T-ALL onset in our experimental model, we performed gene set enrichment analysis (GSEA) to compare gene expression profiles of low and high Trib2 expression from a database (GSE13159) derived from 174 T-ALL patient samples (Figure 6a) [42]. GSEA analysis showed these two groups, defined by Trib2 expression, were of distinct molecular subtypes of T-ALL (Figure 6b). Low Trib2-expressing T-ALL group was enriched with gene set associated with TLX1+ T-ALL (cortical mature T-ALL), whereas high Trib2-expressing T-ALL group had upregulation of LYL1+ T-ALL (early immature T-ALL) gene set [43]. TAL1+ T-ALL gene set was not enriched in either group. A significant positive correlation between Trib2 and Lyl1 expressions was confirmed in an independent T-ALL data set (GSE33315) [44] (Figure 6c). In our experimental model, Trib2−/− T-ALL leukemic cells exhibited deficiencies in MAPK signaling. In accordance with this, using GSEA analysis of low and high Trib2 expressed human T-ALL groups, MAPK signaling was found to be upregulated in the high Trib2-expressing T-ALL group (Figure 6d). Hence, impaired activation of MAPK signaling in the absence of TRIB2 contributed to the increased aggressiveness of NOTCH1-induced murine T-ALL disease that recapitulated the immunophenotypes of human cortical mature T-ALL. Our analysis suggests that TRIB2 possesses tumor-suppressive functions important for T-ALL.

Trib2 levels distinguish molecular subtypes of human T-ALL and associate with MAPK signaling. (a) GSEA was performed to compare human T-ALL samples from GSE13159 data set [42] that expressed low and high Trib2 (n=26 per group) for enrichment of human T-ALL molecular subtypes (b) and canonical pathways (d). NES, normalized enrichment score; Nom, nominal. (c) Correlation between Trib2 (average of 202478_at and 202479_s_at) and Lyl1 (210044_s_at) expressions were examined in 84 human T-ALL samples from GSE33315 data set [44]. For c, Pearson correlation test was used to examination association between Trib2 and Lyl1 expressions.
Discussion
Our data provide strong evidence that TRIB2 is a novel regulator of thymopoietic proliferation, important in the response to stress and the pathogenesis of T-ALL. We have established a previously unrecognized role for TRIB2 as a novel regulator for cellular proliferation of developing thymocytes. At steady state, Trib2−/− developing thymocytes proliferated faster and gave rise to more mature thymocytes. The accelerated proliferation of Trib2−/− developing thymocytes conferred hypersensitivity to 5-FU-induced cell death. Following genotoxic insult, Trib2−/− mice exhibited accelerated thymopoietic recovery due to expansion of c-Kit− DN1 progenitors and high cell division kinetics of developing thymocytes. In the absence of TRIB2, NOTCH1 is more potent in driving T-ALL initiation and the increased aggressiveness of Trib2−/− T-ALL was enhanced by impaired activation of MAPK signaling. Human T-ALL can be distinguished based on Trib2 expression levels and, in accordance with our experimental data, correlate with MAPK signaling in high Trib2-expressing T-ALLs.
Here we show TRIB2 as the first member of the mammalian Tribbles family to have a proliferative role in the context of developing thymocytes during T-cell development. TRIB1 was reported previously to negatively regulate proliferation of human aortic smooth muscle cells in vitro via interaction with MKK4 [45]. TRIB2 regulates cell cycle entry/exit and hence the cell division kinetics of developing thymocytes. We found that the dysregulated proliferation of Trib2−/− developing thymocytes does not affect Tcrb rearrangement, a key event in early T-cell differentiation. Furthermore, loss of TRIB2 did not affect terminal maturation of developing thymocytes. Intriguingly, the increased proliferation of developing thymocytes, in the absence of TRIB2, does not affect the percentage of peripheral T cells. Previous literature has shown that mice overexpressing CD69 have increased thymic SP subsets but a reduction in the number of T cells in the peripheral lymphoid organs [46]. Our data suggest that TRIB2 may impact on thymic selection (DPsm to CD69+ DP transition) or thymic T-cell export. It is also likely that TRIB2 has a distinct role in mature T-cell biology and function because Trib2 expression distinguishes CD4+ from CD8+ peripheral T-cells [23]. This could be further explored using appropriate infection and immunization models.
Thymopoietic recovery is critical to replenish the T-cell repertoire in order to reconstitute cellular immunity due to T-cell depletion in the clinical setting. At steady state, human T-cell repertoire is established during early childhood as removal of the thymus in children after 6 months of age does not cause overt immunodeficiency [47]. However, many medical conditions such as chemo- and irradiation therapy, infection and graft versus host disease can cause T-cell depletion, thymus insult and thus impaired T-cell immunity in patients [48]. Our study showed that TRIB2 limits the thymopoietic recovery after 5-FU injury. The reason underlying this is unclear but tight regulation of thymopoietic restoration has been proposed to prevent adverse consequences such as leukemic transformation of T-cells or autoimmunity [48].
We showed that TRIB2 functions to suppress T-cell leukemogenesis induced by NOTCH1 overexpression. A role for TRIB2 in T-ALL maintenance was demonstrated by Sanda et al [13] , as knockdown of Trib2 in a panel of TAL1-positive human T-ALL cell lines induced apoptosis and inhibited cell growth. This appears to be consistent with the role of TRIB2 in other solid [49–51] and hematological [9] malignancies that overexpression of TRIB2 confers growth and survival advantages to tumor cells. However, we provide strong evidence that TRIB2 has opposing roles in the initiation and potency of T-ALL. In the absence of TRIB2, the latency of NOTCH1-induced murine T-ALL was shortened significantly. We previously reported elevated Trib2 levels in a cohort of pediatric T-ALLs that contain NOTCH1/FBXW7 mutations compared with WT T-ALLs [11]. However, T-ALL is a heterogeneous disease and can be classified into different molecular subtypes, although aberrant NOTCH1 signaling is the unifying feature in all the subtypes. Hence, we performed GSEA analysis in this study to show Trib2 is high in early immature (LYL1+) T-ALLs that are arrested at the DN stage of thymocyte development and show a transcriptional program related to HSCs and myeloid progenitors [44, 52]. In turn, Trib2 is low in TLX1+ human T-ALLs that have a mature cortical DP phenotype where the transformed cells have committed to the T-cell lineage [43]. We propose that the role of TRIB2 in different subtypes of human T-ALL depends on the stage at which thymic progenitors undergo malignant transformation, and if they have committed to T-cell lineage.
There is a strong correlation with Trib2 and MAPK signaling in human T-ALL as shown by GSEA analysis. Previously, the activation of ERK and p38 was shown to inversely correlate with aggressiveness of T-ALL in a model of T-ALL cell dormancy [53]. Furthermore, drug treatment of established human T-ALL cell lines was shown to induce cell death via activation of p38 and JNK [54–56]. Thus, activation of MAPK signaling appears to be a limiting factor, at least, in the context of T-ALL. TRIB family members act as scaffold proteins for the binding of MAPK signaling complexes and hence modulate their activity. All TRIB family members have the conserved MEK1-binding motif and have been shown to interact with MEK1 [38]. In human T-ALLs, upregulation of ERK1/2 signaling was found in 38% of T-ALL patients [57] and MEK/ERK can be activated due to RAS mutations, found in ~10% of T-ALL patients [58]. Targeting of MEK/ERK signaling by MEK inhibition was shown to be effective in treating KRAS-mutated T-ALL in a murine model [59]. The upregulation of MAPK signaling in specific subtypes of T-ALL has not been studied and remains unclear. Here we show that Trib2 expression is high in LYL1+ immature T-ALLs enriched with MAPK signaling, and our experimental data show defective MAPK signaling in the absence of TRIB2. Thus, it appears that TRIB2 functions to control T-ALL via MAPK modulation.
We and others [7, 60] have shown that Trib2 is a myeloid oncogene when overexpressed in a murine BM transplant model. Here loss of TRIB2 potentiated murine T-ALL induced by a T-cell oncogene. Given that Trib2 is expressed highest in normal T cells [19], it is not surprising that overexpression of Trib2 does not drive T-ALL in the BM transplant model, and our data would suggest that TRIB2 functions to suppress T-ALL. Our data provide insight into the understanding of the opposing leukemogenic roles of TRIB2 in myeloid and lymphoid leukemia. However, the mechanisms that underlie whether TRIB2 acts as an oncogene or tumor suppressor are not well understood. It may be linked to its function in cell proliferation and MAPK pathway modulation, which could be cell context specific. Anti- and pro-proliferative effects connected with MAPK signaling and TRIB1 have been previously demonstrated in different cell contexts [38, 45].
To conclude, our studies have shown that TRIB2 is a novel regulator for the cellular proliferation of developing thymocytes, and the response to genotoxic and oncogenic stress. Although the loss of TRIB2 resulted in hypersensitivity to genotoxic stress, thymocytes recovered faster due to the inherent increased proliferation in the absence of TRIB2. These characteristics however are pro-oncogenic, as accelerated T-ALL was observed in the absence of TRIB2. Our findings provide a novel link between proliferative stress and TRIB2 in developing thymocytes, and may prove relevant for clinical practice in hematology and oncology.
Materials and Methods
Antibodies
The following antibodies, from BioLegend (San Diego, CA, USA) and eBioscience (San Diego, CA, USA), were used to stain cells for flow cytometry analysis: anti-B220 (RA3–6B2), anti-BrdU (BU20A), anti-CD3 (17A2), anti-CD4 (RM4–5 and GK1.5), anti-CD8α (53-6.7), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD16/CD32 (93), anti-CD19 (eBio1D3), anti-CD25 (PC61.5), anti-CD34 (RAM34), anti-CD44 (IM7), anti-CD45.2 (104), anti-CD48 (HM48-1), anti-CD71 (R17217) anti-CD150 (TC15-12F12.2), anti-c-Kit (2B8), anti-Gr-1 (RB6–8C5), anti-IL-7Rα (A7R34), anti-Ki-67 (SolA15), anti-NK1.1 (PK136), anti-Sca-1 (D7) and anti-Ter119 (TER-119).
The following antibodies were used for western blotting: anti-p38 (D13E1), anti-phospho-p38 (D3F9), anti-p44/42 (137F5), anti-phospho-p44/42 (D13.14.4E), anti-JNK1 (2C6), anti-phopho-JNK and anti-β-actin (AC-15). Antibodies used to immunoblot MAPK signaling were from Cell Signaling Technology (Danvers, MA, USA) and anti-β-actin antibody was from Sigma-Aldrich (St Louis, MO, USA).
Mice
Trib2−/− (B6; 129S5-Trib2tm1Lex) mice, backcrossed onto C57BL/6, were bred and housed in the biological service unit of the University of Glasgow. Mice (B6.SJL-Ptprca Pepcb/ BoyJ) used in BM transplantation as recipients were bred and housed in the Beatson Biological Service and Research Units. Age-matched (6–14 weeks old) mice of both genders were used for comparison between WT and Trib2−/− genotypes.
Blood cell counts
Mice were sacrificed by carbon dioxide inhalation and blood was subsequently collected by cardiac puncture. Collected blood was analyzed by Hemavet 950 (Drew Scientific, Miami Lakes, FL, USA) to generate complete blood counts and white blood cell differential counts.
Cell preparation, surface staining and flow cytometry
Pelvises, femurs and tibias collected from euthanized mice were dissected free of muscle tissues and tendons. The bones were crushed in 2% fetal bovine serum supplemented phosphate-buffered saline (PBS) solution using a mortar and pestle. The resulting cell suspension was filtered through a cell strainer. Thymus was isolated and grinded directly onto a cell strainer to generate single-cell suspensions. Red blood cell in blood, BM and thymus cell suspensions were lysed by homemade Ammonium-Chloride-Potassium Lysing Buffer containing 10 mm KHCO3, 150 mm NH4Cl and 0.1 mm EDTA. Live-cell count for BM and thymus was then performed by trypan blue exclusion.
Lineage antibody cocktail for BM cell surface staining included anti-CD3, anti-CD4, anti-CD8, anti-B220, anti-CD11b, anti-Gr-1 and anti-Ter119 antibodies. For thymus, non-T-lineage antibody cocktail included anti-CD11c, anti-B220, anti-Gr-1, anti-NK1.1 and anti-Ter119 antibodies. Antibodies used for flow cytometry were conjugated either with single fluorochrome or with biotin and revealed by fluorochrome-labeled streptavidin. All the cell populations identified by flow cytometry were defined in the related figure legends.
Unless indicated otherwise, antibody staining and washing of single-cell suspensions were done in 2% fetal bovine serum supplemented PBS solution. UltraComp eBeads (eBioscience) was used to prepare single-color compensation controls in multicolor flow cytometry experiments. Flow cytometry was performed using BD FACSCanto II system, equipped with blue, red and violet lasers (BD Biosciences, San Jose, CA, USA). Aggregates were excluded by forward scatter (FSC) height versus area signals. The singlet population was then gated by FSC area versus side scatter-area signals to exclude dead cells and debris. Whenever possible, dead cells were also excluded by 4′, 6-diamidino-2-phenylindole or propidium iodide staining. FlowJo (Tree Star, Ashland, OR, USA), was used to analyze flow cytometry data. Fluorescence Minus One controls were used to facilitate gating as appropriate.
In vivo 5-FU treatment
On the drug administration day, mice were weighed to calculate the weight-adjusted dosage, as indicated in the related figure legends, and FU 50 mg ml−1 Solution (Accord Healthcare Limited, North Harrow, Middlesex, UK) was pre-diluted with PBS solution and was administered by intraperitoneal injection.
Apoptosis assay
Thymus was collected from euthanized mice after the indicated period of 5-FU treatment and single-cell suspension was prepared as described above. Cells were stained with Annexin V-fluorescein isothiocyanate and 4’, 6-diamidino-2-phenylindole in Hank’s balanced salt solution, and analyzed by flow cytometry.
Detection of endogenous Tcrb rearrangements
Total thymocytes were digested overnight with proteinase K in published lysis buffer [61] at 56 °C. Samples were then heated at 95 °C for 30 min to inactivate proteinase K. DNA was purified further by phenol extraction and ethanol precipitation. V(D)J rearrangements of the Tcrb locus were amplified by FastStart High Fidelity PCR system (Roche, Indianapolis, IN, USA), with 25 ng of DNA input, using published primers [62]. Primer sequences and the annealing temperatures for each amplification were listed in Supplementary Table S3. PCR products were separated on 1.5% agarose gel, detected by SafeView Nucleic Acid Stain (NBS Biologicals, Huntingdon, Cambridgeshire, UK), and imaged using ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA).
Intracellular staining of Ki-67 and cell cycle analysis
Thymocytes were stained with surface markers to allow subset identification, and then fixed in 4% weight/volume paraformaldehyde-PBS solution for 20 min at room temperature, permeabilized in 0.1% (weight/volume) saponin-PBS solution for 15 min at room temperature, followed by staining with anti-Ki-67 antibody in the permeabilization solution and analyzed by flow cytometry.
To distinguish resting (G0) from active (G1–S/G2/M) cell cycle, surface stained thymocytes were fixed and permeabilized by using BD Cytofix/Cytoperm Kit (BD Biosciences), followed by staining with anti-Ki-67 antibody and DNA selective Vybrant DyeCycle Violet Stain (Life Technologies, Eugene, OR, USA) and analyzed by flow cytometry.
In vivo BrdU pulsing and detection of incorporated BrdU
On day 14 post 5-FU treatment, mice were pulsed with 1 mg BrdU-labeling reagent (Invitrogen, Camarillo, CA, USA), by intraperitoneal injection, for 1 or 4 h. Thymus was collected from euthanized mice and single-cell suspension was prepared. Surface-stained thymocytes were fixed in BD Cytofix/Cytoperm Buffer for 30 min on ice and stored at −80 °C for overnight in 10% dimethylsulfoxide supplemented fetal bovine serum to permeabilize the nuclear membrane. Cells were then thawed at 37 °C, fixed with BD Cytofix/Cytoperm Buffer for an additional 10 min on ice and treated with DNase I (30 μl per 1×106 cells) from STEMCELL Technologies (Vancouver, BC, Canada) for 1 h at 37 °C. Cells were stained with anti-BrdU antibody and Vybrant DyeCycle Violet Stain (Life Technologies) in BD Perm/Wash Buffer (San Jose, CA, USA) and analyzed by flow cytometry.
Bone marrow transduction and transplantation
Retroviral transduction of WT and Trib2−/− BM cells, and subsequent transfer of these cells into lethally irradiated recipients (B6.SJL) were performed as described previously [7]. In brief, BM cells were collected from 6–8-week-old mice 4 days after administration of 5-FU (250 mg kg–1, intraperitoneally) and retrovirally transduced ex vivo, with MigR1 or MigR1-ICN1, in the presence of interleukin (IL)-3 (10 ng ml−1), IL-6 (10 ng ml−1) and stem cell factor (100 ng ml−1). Retroviral supernatants for MigR1 and MigR1-ICN1 were generated by transient transfection of 293T cells (together with pCMV-Gag-Pol packaging vector and pCMV-VSV-G envelope vector) and titered with 3T3 cell as previously described [10]. MigR1 and MigR1-ICN1 retroviral supernatants with equal titers were used to ensure similar transduction efficiency across three independent transplant experiments. Cells (0.25–0.5×106) were then injected intravenously into lethally irradiated (2×4.25 grays fractionated doses were given 3 h apart) recipients. Mice were monitored by periodic tail vein bleedings 3 weeks after transplantation. Mice were sacrificed when they showed any of the following symptoms of disease: severe cachexia, lethargy, white blood cell count >20×106 cells per ml and hunching. Healthy controls were sacrificed at the end of experiment when all mice in the test groups succumbed to disease.
GSEA analysis
Gene expression profiles of T-ALL and healthy BM samples from GSE13159 data set [42] were downloaded from the Leukemia Gene Atlas platform [63]. On the basis of 202478_at feature, Trib2 expression of low Trib2 T-ALL group ranged from 3.602 to 5.370, whereas high Trib2 T-ALL group ranged from 8.811 to 11.103. The mean value of Trib2 for healthy BM samples (n=73) was 6.172. GSEA [64] was performed using the default settings. Gene sets from the Molecular Signatures Database v5.0 of GSEA (Broad Institute of MIT and Harvard, Cambridge, MA, USA) were used. These included the 1330 gene sets in C2: CP collection for canonical pathways and the three gene sets in C2: CGP collection (M4175, M2059 and M1007) for T-ALL molecular subtypes.
Western blotting
Total BM cells collected from euthanized mice were lysed in homemade Radio-Immunoprecipitation Assay buffer (50 mm Tris buffer (pH7.4), 150 mm NaCl, 0.5% igepal CA 630, 0.25% sodium deoxycholate and 1 mm EDTA). Protein lysates were resolved by sodium dodecylsulfate polyacrylamide gel electrophoresis using 12% gels and transferred to 0.45-μm nitrocellulose membranes. The membranes were probed with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies. Stripping buffer (Thermo Scientific, Rockford, IL, USA) was used as appropriate for secondary probing.
Statistics
GraphPad Prism (version 5.03; La Jolla, CA, USA) was used for statistical analysis and graphing. An unpaired, two-tailed, Student’s t-test was applied for comparison of two groups. For analysis of multiple groups between WT and Trib2−/− genotypes, two-way analysis of variance with Bonferroni post tests was applied. Log-rank test was applied for survival curve comparison. Pearson correlation test was applied to examine association between Trib2 and Lyl1 expressions of T-ALL samples from GSE33315 data set [44]. This data set was downloaded from Gene Expression Omnibus and Robust Multichip Average method was used for data normalization. D’Agostino-Pearson omnibus test confirmed the T-ALL samples had normal distributions of Trib2 and Lyl1 values. Statistical significance of differences was attained when P-value <0.05 and was indicated in the related graphs.
Study approval
All mouse experiments were approved by UK Animal Ethical Committees and performed according to UK Home Office project license (Animal (Scientific Procedures) Act 1986) guidelines.
References
Yokoyama T, Nakamura T . Tribbles in disease: Signaling pathways important for cellular function and neoplastic transformation. Cancer Sci 2011; 102: 1115–1122.
Saka Y, Smith JC . A Xenopus tribbles orthologue is required for the progression of mitosis and for development of the nervous system. Dev Biol 2004; 273: 210–225.
Grosshans J, Wieschaus E . A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell 2000; 101: 523–531.
Mata J, Curado S, Ephrussi A, Rorth P . Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell 2000; 101: 511–522.
Seher TC, Leptin M . Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr Biol 2000; 10: 623–629.
Takasato M, Kobayashi C, Okabayashi K et al. Trb2, a mouse homolog of tribbles, is dispensable for kidney and mouse development. Biochem Biophys Res Commun 2008; 373: 648–652.
Keeshan K, He Y, Wouters BJ et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell 2006; 10: 401–411.
Argiropoulos B, Palmqvist L, Yung E et al. Linkage of Meis1 leukemogenic activity to multiple downstream effectors including Trib2 and Ccl3. Exp Hematol 2008; 36: 845–859.
Rishi L, Hannon M, Salome M et al. Regulation of Trib2 by an E2F1-C/EBPalpha feedback loop in AML cell proliferation. Blood 2014; 123: 2389–2400.
Wouters BJ, Jorda MA, Keeshan K et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood 2007; 110: 3706–3714.
Hannon MM, Lohan F, Erbilgin Y et al. Elevated TRIB2 with NOTCH1 activation in paediatric/adult T-ALL. Br J Haematol 2012; 158: 626–634.
Nagel S, Venturini L, Przybylski GK et al. Activation of Paired-homeobox gene PITX1 by del(5)(q31) in T-cell acute lymphoblastic leukemia. Leuk Lymphoma 2011; 52: 1348–1359.
Sanda T, Lawton LN, Barrasa MI et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 2012; 22: 209–221.
Li FX, Zhu JW, Hogan CJ, DeGregori J . Defective gene expression, S phase progression, and maturation during hematopoiesis in E2F1/E2F2 mutant mice. Mol Cell Biol 2003; 23: 3607–3622.
Zeddies S, Jansen SB, di Summa F et al. MEIS1 regulates early erythroid and megakaryocytic cell fate. Haematologica 2014; 99: 1555–1564.
Ariki R, Morikawa S, Mabuchi Y et al. Homeodomain transcription factor Meis1 is a critical regulator of adult bone marrow hematopoiesis. PLoS ONE 2014; 9: e87646.
Tanigaki K, Honjo T . Regulation of lymphocyte development by Notch signaling. Nat Immunol 2007; 8: 451–456.
Gekas C, Rhodes KE, Gereige LM et al. Mef2C is a lineage-restricted target of Scl/Tal1 and regulates megakaryopoiesis and B-cell homeostasis. Blood 2009; 113: 3461–3471.
Liang KL, Rishi L, Keeshan K . Tribbles in acute leukemia. Blood 2013; 121: 4265–4270.
Bhandoola A, Sambandam A . From stem cell to T cell: one route or many? Nat Rev Immunol 2006; 6: 117–126.
Hoffman ES, Passoni L, Crompton T et al. Productive T-cell receptor beta-chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev 1996; 10: 948–962.
Taghon T, Yui MA, Pant R, Diamond RA, Rothenberg EV . Developmental and molecular characterization of emerging beta- and gammadelta-selected pre-T cells in the adult mouse thymus. Immunity 2006; 24: 53–64.
Mingueneau M, Kreslavsky T, Gray D et al. The transcriptional landscape of alphabeta T cell differentiation. Nat Immunol 2013; 14: 619–632.
Seitan VC, Hao B, Tachibana-Konwalski K et al. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 2011; 476: 467–471.
Brekelmans P, van Soest P, Voerman J, Platenburg PP, Leenen PJ, van Ewijk W . Transferrin receptor expression as a marker of immature cycling thymocytes in the mouse. Cell Immunol 1994; 159: 331–339.
Porritt HE, Rumfelt LL, Tabrizifard S, Schmitt TM, Zuniga-Pflucker JC, Petrie HT . Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity 2004; 20: 735–745.
Byeon IJ, Li H, Song H, Gronenborn AM, Tsai MD . Sequential phosphorylation and multisite interactions characterize specific target recognition by the FHA domain of Ki67. Nat Struct Mol Biol 2005; 12: 987–993.
Li Z, Dordai DI, Lee J, Desiderio S . A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity 1996; 5: 575–589.
Lin WC, Desiderio S . Cell cycle regulation of V(D)J recombination-activating protein RAG-2. Proc Natl Acad Sci USA 1994; 91: 2733–2737.
Longley DB, Harkin DP, Johnston PG . 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3: 330–338.
Eichhorst ST, Muerkoster S, Weigand MA, Krammer PH . The chemotherapeutic drug 5-fluorouracil induces apoptosis in mouse thymocytes in vivo via activation of the CD95(APO-1/Fas) system. Cancer Res 2001; 61: 243–248.
Shin JY, Hu W, Naramura M, Park CY . High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J Exp Med 2014; 211: 217–231.
Aquino Esperanza JA, Aguirre MV, Aispuru GR et al. In vivo 5-fluorouracil-[corrected]induced apoptosis on murine thymocytes: involvement of FAS, Bax and Caspase3. Cell Biol Toxicol 2008; 24: 411–422.
Anderson G, Takahama Y . Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol 2012; 33: 256–263.
Benz C, Martins VC, Radtke F, Bleul CC . The stream of precursors that colonizes the thymus proceeds selectively through the early T lineage precursor stage of T cell development. J Exp Med 2008; 205: 1187–1199.
Kondo M, Weissman IL, Akashi K . Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997; 91: 661–672.
Aster JC, Xu L, Karnell FG, Patriub V, Pui JC, Pear WS . Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol Cell Biol 2000; 20: 7505–7515.
Yokoyama T, Kanno Y, Yamazaki Y, Takahara T, Miyata S, Nakamura T . Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood 2010; 116: 2768–2775.
Wang PY, Sun YX, Zhang S et al. Let-7c inhibits A549 cell proliferation through oncogenic TRIB2 related factors. FEBS Lett 2013; 587: 2675–2681.
Wei SC, Rosenberg IM, Cao Z, Huett AS, Xavier RJ, Podolsky DK . Tribbles 2 (Trib2) is a novel regulator of toll-like receptor 5 signaling. Inflamm Bowel Dis 2012; 18: 877–888.
Izrailit J, Berman HK, Datti A, Wrana JL, Reedijk M . High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/TGFbeta pathways as fundamental Notch regulators in breast cancer. Proc Natl Acad Sci USA 2013; 110: 1714–1719.
Haferlach T, Kohlmann A, Wieczorek L et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 2010; 28: 2529–2537.
Ferrando AA, Neuberg DS, Staunton J et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1: 75–87.
Zhang J, Ding L, Holmfeldt L et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012; 481: 157–163.
Sung HY, Guan H, Czibula A et al. Human tribbles-1 controls proliferation and chemotaxis of smooth muscle cells via MAPK signaling pathways. J Biol Chem 2007; 282: 18379–18387.
Nakayama T, Kasprowicz DJ, Yamashita M et al. The generation of mature, single-positive thymocytes in vivo is dysregulated by CD69 blockade or overexpression. J Immunol 2002; 168: 87–94.
Spits H . Development of alphabeta T cells in the human thymus. Nat Rev Immunol 2002; 2: 760–772.
Boehm T, Swann JB . Thymus involution and regeneration: two sides of the same coin? Nat Rev Immunol 2013; 13: 831–838.
Zanella F, Renner O, Garcia B et al. Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells. Oncogene 2010; 29: 2973–2982.
Grandinetti KB, Stevens TA, Ha S et al. Overexpression of TRIB2 in human lung cancers contributes to tumorigenesis through downregulation of C/EBPalpha. Oncogene 2011; 30: 3328–3335.
Wang J, Park JS, Wei Y et al. TRIB2 acts downstream of Wnt/TCF in liver cancer cells to regulate YAP and C/EBPalpha function. Mol Cell 2013; 51: 211–225.
Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood 2013; 122: 74–82.
Masiero M, Minuzzo S, Pusceddu I et al. Notch3-mediated regulation of MKP-1 levels promotes survival of T acute lymphoblastic leukemia cells. Leukemia 2011; 25: 588–598.
Liu Y, Ge J, Li Q et al. Low-dose anisomycin sensitizes glucocorticoid-resistant T-acute lymphoblastic leukemia CEM-C1 cells to dexamethasone-induced apoptosis through activation of glucocorticoid receptor and p38-MAPK/JNK. Leuk Lymphoma 2014; 55: 2179–2188.
Ge J, Liu Y, Li Q et al. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed Environ Sci 2013; 26: 902–911.
Jiang Q, Li F, Shi K et al. ATF4 activation by the p38MAPK-eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett 2013; 587: 2420–2429.
Gregorj C, Ricciardi MR, Petrucci MT et al. ERK1/2 phosphorylation is an independent predictor of complete remission in newly diagnosed adult acute lymphoblastic leukemia. Blood 2007; 109: 5473–5476.
Kindler T, Cornejo MG, Scholl C et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood 2008; 112: 3373–3382.
Dail M, Li Q, McDaniel A et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci USA 2010; 107: 5106–5111.
Dedhia PH, Keeshan K, Uljon S et al. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood 2010; 116: 1321–1328.
Wang X, Xiao G, Zhang Y et al. Regulation of Tcrb recombination ordering by c-Fos-dependent RAG deposition. Nat Immunol 2008; 9: 794–801.
Cobaleda C, Jochum W, Busslinger M . Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 2007; 449: 473–477.
Hebestreit K, Grottrup S, Emden D et al. Leukemia gene atlas--a public platform for integrative exploration of genome-wide molecular data. PLoS ONE 2012; 7: e39148.
Subramanian A, Tamayo P, Mootha VK et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Acknowledgements
We thank Ashley Newcombe, Hothri Ananyamb Moka, Joana Campos, Karen Dunn and all the technical staff at the Paul O’Gorman Leukemia Research Centre. We thank the Cancer Research UK Glasgow Centre (C596/A18076) and the Biological Service Unit facilities at the Cancer Research UK Beatson Institute (C596/A17196) and the Biological Services at the University of Glasgow. We thank Alison Michie for helpful suggestions. We thank the Kay Kendall foundation (KKL501), and the Howat foundation for flow cytometry facility funding. KK was supported by the Howat Foundation and Children with Cancer UK. KL was supported by Health Research Board Ireland (PhD Scholars Program in Cancer Biology). CO’C was supported by Children’s Leukemia Research Project. PV was supported by the Leukaemia & Lymphoma Research Project Grant 13011.
Author information
Authors and Affiliations
Contributions
KK and KL designed the study, analyzed the data and wrote the paper. KL, CO’C and PV performed the research. TVM supervised and guided the study. KL made the figures. All authors edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Discovery website.)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liang, K., O’Connor, C., Veiga, J. et al. TRIB2 regulates normal and stress-induced thymocyte proliferation. Cell Discov 2, 15050 (2016). https://doi.org/10.1038/celldisc.2015.50
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/celldisc.2015.50
This article is cited by
-
A miRNAs catalogue from third-stage larvae and extracellular vesicles of Anisakis pegreffii provides new clues for host-parasite interplay
Scientific Reports (2022)
-
Tribbles homolog 2 (Trib2), a pseudo serine/threonine kinase in tumorigenesis and stem cell fate decisions
Cell Communication and Signaling (2021)
-
TRIB3 promotes MYC-associated lymphoma development through suppression of UBE3B-mediated MYC degradation
Nature Communications (2020)
-
Oncogenic transcriptional program driven by TAL1 in T-cell acute lymphoblastic leukemia
International Journal of Hematology (2019)
-
A Trib2-p38 axis controls myeloid leukaemia cell cycle and stress response signalling
Cell Death & Disease (2018)
