
Abstract
12/15-Lipoxygenase (12/15-LO) is an enzyme that converts polyunsaturated fatty acids into bioactive lipid derivatives. In this study, we showed that inhibition of 12/15-LO by baicalein (BA) significantly attenuated clinical severity of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS). Inhibited migration of autoimmune T cells into the central nervous system (CNS) by BA treatment could be attributed to reduced activation of microglia, which was indicated by suppressed phagocytosis, and decreased production of proinflammatory cytokines and chemokines in the CNS. We further observed that inhibition of 12/15-LO with BA led to increased expression of peroxisome proliferator-activated receptor (PPAR)β/δ in microglia of EAE mice. This was confirmed in vitro in primary microglia and a microglia cell line, BV2. In addition, we demonstrated that BA did not affect 12/15-LO or 5-lipoxygenase (5-LO) expression in microglia, but significantly decreased 12/15-LO products without influencing the levels of 5-LO metabolites. Moreover, among these compounds only 12/15-LO metabolite 12-hydroxyeicosatetraenoic acid was able to reverse BA-mediated upregulation of PPARβ/δ in BV2 cells. We also showed that inhibition of microglia activation by PPARβ/δ was associated with repressed NF-κB and MAPK activities. Our findings indicate that inhibition of 12/15-LO induces PPARβ/δ, demonstrating important regulatory properties of 12/15-LO in CNS inflammation. This reveals potential therapeutic applications for MS.
Similar content being viewed by others


Main
Multiple sclerosis (MS) is an autoimmune, demyelinating disease of the central nervous system (CNS) resulting in motor and sensory deficits. Altered plasma lipid profiles were found in many MS patients,1 suggesting a role of lipids in pathogenesis of MS. For example, in active MS, there is a shift in the lipid composition of normal appearing white matter and normal appearing grey matter to a higher phospholipid and lower sphingolipid content, which indicates a metabolic defect that causes sphingolipids to be shuttled to phospholipid production in MS patients.2 Experimental autoimmune encephalomyelitis (EAE) is a commonly used animal model for MS. Autoreactive T helper (Th) 1 and Th17 cells responding to myelin proteins are implicated in the pathogenesis of MS and EAE,3, 4 and CNS-activated microglia are thought to participate in myelin loss and lesion formation.5 It is evident that disregulation of lipid metabolism is linked with disease progression of EAE. For example, anandamide is a derivative of arachidonic acid, fatty acid amide hydrolase and epidermal fatty acid-binding protein are involved in the degradation and transportation of anandamide respectively.6 Genetic deletion of fatty acid amide hydrolase resulted in improved long-term outcome in chronic autoimmune encephalitis,7 and mice deficient in epidermal fatty acid-binding protein developed milder EAE.8
12/15-Lipoxygenase (12/15-LO) belongs to a family of enzymes that can metabolize arachidonic acid to generate corresponding hydroxides such as 12-hydroxyeicosatetraenoic acid (12-HETE) and 15-HETE.9, 10 Arachidonic acid and its lipid metabolites have important roles in human inflammatory diseases,11, 12, 13 which suggests that 12/15-LO can be a pathogenic player that mediates inflammatory responses in vivo. 12/15-LO is widely expressed in the CNS,14, 15 and has been demonstrated to be involved in the pathogenesis of various neurological diseases.16, 17, 18, 19 12/15-LO exerts a regulatory role in Alzheimer’s disease, an inflammatory and neurodegenerative disease.16, 17 Li et al.18 demonstrated that decreased neuronal concentrations of the anti-oxidant glutathione, an early biomarker in Parkinson’s disease, was associated with 12/15-LO activation. A recent study reported that 12/15-LO-deficient mice showed a late but severe clinical symptom as compared with wild-type mice.19 However, the authors did not address the underlying cellular and molecular mechanisms directly. Thus, the exact role of 12/15-LO in EAE remains unresolved.
The peroxisome proliferator-activated receptors (PPARs) are a family of three ligand-activated transcription factors: PPARα, PPARβ/δ and PPARγ. All the PPARs have critical roles in regulation of development and energy homeostasis.20 Recently, there has been a great deal of interest in the involvement of PPARs in inflammatory processes.21 In particular, PPARα and PPARγ inhibit the activation of inflammatory gene expression and interfere with proinflammatory transcription factor signaling pathways in CNS inflammatory diseases.22, 23 However, the role of PPARβ/δ for regulating inflammation is just emerging, and the mechanisms by which PPARβ/δ affects inflammatory reactions are not well understood.
Baicalein (BA) is a specific and commonly used inhibitor of leukocyte 12/15-LO.24, 25 In this study, we examined the role of 12/15-LO in regulating inflammatory responses during pathogenesis of EAE. We found that on selective inhibition of 12/15-LO with BA, mice displayed much milder EAE symptoms despite their competent peripheral T-cell response. BA functioned in microglia to promote PPARβ/δ expression, which in turn inhibited microglia activation, reduced production of proinflammatory cytokines and chemokines, and decreased immune cell infiltration into the CNS. These findings indicate a regulatory role of 12/15-LO in inflammation with potential therapeutic benefits in CNS autoimmune diseases.
Results
The 12/15-LO inhibitor BA ameliorates clinical symptoms of EAE
To investigate the role of 12/15-LO in CNS autoimmune disease, we used BA (75 mg/kg/day), a 12/15-LO inhibitor, by i.p. injection in EAE mice from day 3 pre-immunization onward as a preventive protocol. Vehicle-treated mice served as controls. BA treatment significantly attenuated the severity of EAE compared with the vehicle control (Figure 1a). The effects were noticeable at day 13 post immunization and persisted over the entire course of EAE. Similar effects were observed when treatment began from day 12 post-immunization onward as a treatment protocol (Figure 1b). The observed clinical effects of BA were consistent with much less infiltration of inflammatory cells and fewer demyelinated plaques in the white matter of BA-treated EAE mice compared with vehicle-treated group (Figure 1c). Moreover, percentages of inflammatory CD4+ and CD8+ T cells that infiltrated into the CNS of naive, vehicle- and BA-treated EAE mice in induction phase were examined (Figure 1d). We found that BA significantly reduced infiltration of inflammatory T cells into the CNS of EAE mice compared with vehicle control. These data indicate that 12/15-LO inhibitor BA is effective in alleviating the severity of EAE.

The 12/15-LO inhibitor BA ameliorated EAE severity. (a and b) Clinical scores of EAE mice subjected to vehicle or BA treatment with the preventive (a) and treatment (b) protocols. Results are shown as mean±S.E.M. (n=6 mice per group) and representative of three independent experiments with similar results. (c and d) EAE mice were subjected to vehicle or BA treatment with the preventive protocol. Spinal cord sections were stained with H&E (left, top) or LFB (left, bottom) on day 20 post immunization. Original magnification, × 40. Cellular infiltration in H&E sections and demyelinated areas in LFB-stained sections were summarized as mean±S.E.M. (n=6) on the right. Scale bars, 500 μm (c). Flow cytometry were performed to analyze infiltration of CD45+CD4+ and CD45+CD8+ T cells in the spinal cords on day 10 post immunization (d). Results in c (left) and d are representative of three independent experiments. *P<0.05; **P<0.01
BA reduces autoimmune cell infiltration into the CNS
It is known that efficient activation of autoreactive T cells is an obligatory requirement for induction of CNS inflammation and pathology of EAE.26 We tested whether EAE attenuated by BA was the result of inhibition of T-cell proliferation and cytokine production. Draining lymph nodes (DLNs) and splenocytes were isolated from vehicle- or BA-treated EAE mice for analyses. Proliferation of encephalitogenic T cells was comparable between groups on the myelin oligodendrocyte glycoprotein (MOG) peptide stimulation ex vivo (Figure 2a). In addition, BA did not alter the capability of encephalitogenic T cells to produce cytokines such as interferon (IFN)-γ, interleukin (IL)-4, IL-10 and IL-17, as compared with vehicle control (Figure 2b).

BA reduced autoimmune cell infiltration into the CNS without impairing immune responses in the periphery. (a–c and e–g) EAE mice were subjected to vehicle or BA treatment with the preventive protocol, 10 days after MOG peptide immunization, DLNs, spleens and spinal cords were isolated for further experiments. (a) DLN cells and splenocytes were re-challenged with the MOG peptide at the indicated concentrations for 3 days and examined for proliferation. Data are shown as mean±S.E.M. (n=6). (b) Culture supernatants derived from cells re-challenged with 20 μg/ml of MOG peptide for 48 h were collected for cytokine measurement by ELISA. Results in a and b are shown as mean±S.E.M. (n=6). (c) DLN cells and splenocytes re-challenged with 10 μg/ml of MOG peptide for 3 days were transferred into sublethally irradiated mice (6 Gy). Recipients were monitored and scored daily. (d) DLN cells and splenocytes were isolated from EAE mice on day 10 and re-challenged with 10 μg/ml of MOG peptide for 3 days and were transferred into sublethally irradiated mice to induce EAE. Recipients were treated with vehicle or BA (75 mg/kg/day) from day 7 post-transfer onward. Clinical scores were monitored daily. Results in c and d are shown as mean±S.E.M. (n=6 mice per group) and representative of three independent experiments with similar results. (e and f) Real-time PCR analyses of mRNAs of chemokines and cytokines in the spinal cords. Data are normalized to the gene expression in vehicle-treated mice and shown as mean±S.E.M. (n=6). (g) Flow cytometry analyses of CXCR3 and CCR6 expression on CD4+ T cells in the spinal cords and DLNs. Results are representative of three independent experiments with similar results. *P<0.05; **P<0.01
To further examine the effects of BA on periphery autoimmune cell responses, DLN cells and splenocytes derived from vehicle- or BA-treated EAE mice were isolated, re-stimulated with the MOG peptide in vitro, and adoptively transferred into sublethally irradiated mice. Lymphocytes derived from both groups were able to induce EAE without significant difference (Figure 2c). In another study, DLN cells and splenocytes from EAE mice were re-stimulated with the MOG peptide in vitro and adoptively transferred into sublethally irradiated mice. These mice then received vehicle or BA treatment from day 7 post transfer. Notably, BA significantly suppressed EAE compared with vehicle control (Figure 2d). This suggests a possible effect of BA on migration of inflammatory cells into the CNS. To test this, messenger RNAs (mRNAs) encoding inflammation-associated chemokines such as CCL2, CCL3, CCL20 and CXCL10 were examined. BA treatment significantly reduced expression of chemokines in the CNS (Figure 2e). In addition, expression of the Th1 and Th17 cytokines, IFN-γ and IL-17, was reduced in the CNS of BA-treated EAE mice (Figure 2f). CXCR3 and CCR6, the signature chemokine receptors of Th1 and Th17 cells, were also examined by flow cytometry of CD4+ T cells derived from the CNS and DLNs. The percentages of CXCR3+CD4+ and CCR6+CD4+ cells were significantly lower in the CNS, but higher in DLN cells of BA-treated EAE mice compared with control mice (Figure 2g). These results suggest that BA treatment leads to impaired Th1 and Th17 cell migration into the CNS and relative accumulation of these cells in DLNs. Thus, BA treatment ameliorates clinical symptoms of EAE through inhibition of migration of autoimmune T cells into the CNS.
BA suppresses CNS inflammation through inhibition of microglia activation
Microglia and astrocytes are CNS-resident cells that have important roles in EAE pathogenesis.27, 28, 29, 30 On activation, these cells produce proinflammatory factors that are toxic to the CNS and promote infiltration and reactivation of autoimmune cells in the CNS. In this regard, we examined the expression of 12/15-LO in these two cell populations on BA treatment. 12/15-LO was mainly expressed in murine primary microglia rather than astrocytes at both the mRNA and protein levels (Figures 3a and b). BA treatment did not affect the expression of 12/15-LO in primary microglia, astrocytes or microglia derived from EAE mice (Figures 3a–c), but significantly reduced 12/15-LO metabolites such as 12-HETE, 13-hydroxyoctadecadienoic acid (13-HODE) and 15-HETE in the culture supernatants of primary microglia (Figure 3d). Another lipoxygenase isoform, 5-lipoxygenase (5-LO), was also found to be expressed in primary microglia, astrocytes and microglia derived from EAE mice (Figures 3a–c). BA affected neither 5-LO expression in these cell populations, nor its metabolites leukotriene B4 (LTB4) or leukotriene D4 (LTD4) in the culture supernatants of primary microglia (Figures 3a–d). These data indicate that BA is able to selectively inhibit 12/15-LO activity in microglia. Electron microscopy (EM) was then used to observe the status of microglia. Figure 3e showed microglia in a demyelinated region from vehicle-treated EAE mice. These cells presented long cisternae of granular endoplasmic reticulum and more irregular cell body shapes and exhibited phagocytosis of destroyed myelin and cellular debris. BA treatment suppressed phagocytosis by microglia, as characterized by fewer fragments in the cytoplasm, suggesting impaired microglia activation.

BA suppressed CNS inflammation through inhibition of microglia activation. (a) Real-time PCR analyses of 12/15-LO (left) and 5-LO (right) expression in primary microglia or astrocytes. Data are normalized to the gene expression in vehicle-treated cells and shown as mean±S.E.M. (n=6). (b) Immunoblot analyses of 12/15-LO and 5-LO in vehicle- or BA-treated primary microglia and astrocytes. (c) CD11b+ cells were isolated from the spinal cords of vehicle- or BA-treated EAE mice on day 10 post immunization. Levels of 12/15-LO and 5-LO were determined by immunoblot analyses. (d) Primary microglia and astrocytes were treated with vehicle or 20 μM BA for 24 h. Levels of 12-HETE, 13-HODE, 15-HETE, LTB4 and LTD4 in the supernatants were measured by ELISA. Results are shown as mean±S.E.M. (n=6). (e) EM images of microglia (M) in spinal cords of vehicle- or BA-treated EAE mice. Original magnification, × 4200. Scale bar, 2.5 μm. (f) Primary microglia were left untreated or treated with vehicle or BA (10 or 20 μM) for 12 h and then stimulated with LPS (100 ng/ml) for 3 h. Expression of proinflammatory cytokines and chemokines were analyzed by real-time PCR. Data are normalized to the gene expression in vehicle-treated microglia without LPS stimulation and shown as mean±S.E.M. (n=6). Results in b, c and e are representative of three independent experiments with similar results. *P<0.05; **P<0.01
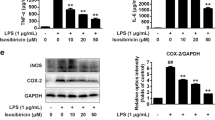
To further elucidate the impact of BA on microglia, we assessed the expression profiles of proinflammatory cytokine and chemokine mRNAs in LPS-stimulated, primary microglia treated with or without BA. Three-hour exposure of microglia to LPS significantly increased expression of proinflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-1β, IL-6 and IL-12p40, and chemokines such as CCL2, CCL3, CCL20 and CXCL10 (Figure 3f). However, pre-treatment with BA before LPS challenge markedly reduced expression of these factors in a dose-dependent manner (Figure 3f). By contrast, BA treatment did not reverse the increased proinflammatory gene expression on IFN-γ stimulation in primary astrocytes (data not shown). Collectively, these data suggest that the anti-inflammatory property of BA in the CNS of EAE mice is mainly due to its modulatory effects on microglia activation.
BA increases PPARβ/δ in microglia
PPARs are key modulators of lipid metabolism and exhibit anti-inflammatory effects in EAE.31 We next investigated expression of PPARs in the CNS and peripheral immune tissues of vehicle- and BA-treated EAE mice. Expression of PPARβ/δ in the CNS was significantly reduced in the induction and peak phases of EAE. Interestingly, BA treatment significantly reversed PPARβ/δ expression within the CNS (Figure 4a), whereas the same treatment did not induce PPARβ/δ expression in the periphery as detected in DLNs (Figure 4b). It is known that PPARγ can be induced through activation of the 12/15-LO pathway, while suppressed on inhibition of 12/15-LO.13, 32 Intriguingly, little PPARγ protein expression was detected in the CNS of naive mice and EAE mice, and BA treatment did not further reduce PPARγ protein levels (Figure 4a). This indicates that BA has only marginal effects on PPARγ expression in the CNS, and it is likely that recovery of PPARβ/δ in the CNS on BA treatment can have an important role in amelioration of EAE.

BA-induced PPARβ/δ in microglia. (a–c) EAE mice were subjected to vehicle or BA treatment with the preventive protocol, spinal cords (a) and DLNs (b) were isolated on day 10 (a representative day of induction phase) and day 20 (a representative day of peak phase) post immunization for immunoblot analyses of PPARβ/δ (a and b) and PPARγ (a). Samples isolated from naive mice were set as controls. (c) CD11b+ cells were isolated from spinal cords on day 10 post immunization for detection of PPARβ/δ by real-time PCR. Data are normalized to the gene expression in vehicle-treated mice and shown as mean±S.E.M. (n=6). (d) Primary microglia treated with vehicle or BA (10 or 20 μM) for 24 h were subjected to immunoblot analyses of PPARβ/δ. (e) Primary microglia were cultured in the absence or presence of vehicle or BA (10 or 20 μM) for 24 h and followed by stimulation with 100 ng/ml of LPS for 3 h, then subjected to immunofluorescence microscopy of CD11b (green) and PPARβ/δ (red). Cells were counter stained with DAPI (blue) to indicate nuclei. Scale bar, 20 μm (top). The fluorescence intensity of PPARβ/δ in nuclei and cytoplasm areas was quantitated using ImageJ software. The relative constituent ratios of PPARβ/δ in nuclei and cytoplasm are shown to indicate the intensity and localization of PPARβ/δ in nuclei and cytoplasm (bottom). Results in a, b, d and e are representative of three independent experiments with similar results. **P<0.01
As we have demonstrated that BA suppressed EAE through inhibition of microglia activation, we then examined the effect of BA on PPARβ/δ expression in microglia. The mRNA level of PPARβ/δ was significantly higher in CD11b+ cells isolated from BA-treated EAE mice compared with control mice (Figure 4c). In addition, BA increased PPARβ/δ protein level in primary microglia in a dose-dependent manner (Figure 4d). Immunofluorescence microscopy showed that the localization of PPARβ/δ was visualized exclusively in cytoplasm in both medium and LPS-treated microglia, whereas pre-treatment with BA led to remarkable accumulation of PPARβ/δ in the nuclei in microglia stimulated with LPS (Figure 4e), indicating increased PPARβ/δ transcriptional activity on BA treatment.
The mechanisms of PPARβ/δ induction in microglia on BA treatment were further explored by using a microglia cell line, BV2. In accordance with the above data, BA significantly increased expression of PPARβ/δ in a dose- and time-dependent manner (Figure 5a). BA did not affect 12/15-LO or 5-LO expression in BV2 cells (Figure 5b), but reduced 12/15-LO metabolites 12-HETE, 13-HODE and 15-HETE without decreasing 5-LO metabolites LTB4 or LTD4 in the culture supernatants (Figure 5c). Moreover, 12-HETE, the main metabolite of 12/15-LO, but not LTB4 or LTD4 markedly reversed upregulation of PPARβ/δ by BA in BV2 cells (Figure 5d). In addition, the effects on PPARβ/δ expression were also analyzed by using another 12/15-LO inhibitor, PD146176, and a 5-LO-selective inhibitor, Zileuton, together with BA. As shown in Figure 5e, BA markedly induced the expression of PPARβ/δ in BV2 cells. PD146176 also contributed to the induction of PPARβ/δ, but to a lesser extent. However, Zileuton did not upregulate the expression of PPARβ/δ. For further examination, siRNA against 12/15-LO was transfected into BV2 cells. On knockdown of 12/15-LO, expression of PPARβ/δ was significantly increased (Figure 5f). As PPARβ/δ is a ligand-dependent nuclear receptor, the binding activity of PPARβ/δ to the peroxisome proliferator DNA-response element with nuclear extracts from cells exposed to BA, PD146176 or Zileuton was examined. Again, BA and PD146176 but not Zileuton induced significantly elevated DNA-binding activities of PPARβ/δ (Figure 5g). Collectively, these observations demonstrate that induction of functional PPARβ/δ by BA is dependent on 12/15-LO inhibition in microglia.

BA-induced PPARβ/δ in microglia through inhibition of 12/15-LO. (a) Immunoblot analyses of PPARβ/δ in BV2 cells treated with BA at indicated concentrations (left) or different time points (right). (b and c) BV2 cells were treated with vehicle or BA treatment for 24 h, cells were then subjected to immunoblot analyses of 12/15-LO and 5-LO (b). Supernatants were collected for analyses of 12-HETE, 13-HODE, 15-HETE, LTB4 and LTD4 by ELISA (c). Results are shown as mean±S.E.M. (n=6). (d) BV2 cells were left untreated or treated with vehicle or 20 μM BA. For cells treated with BA, 12-HETE (100 pM), LTB4 (100 nM) or LTD4 (100 nM) was included in the culture. After 24 h, cells were harvested for analyses of PPARβ/δ by immunoblots. (e) Immunoblot analyses of PPARβ/δ in BV2 cells subjected to vehicle, Zileuton (10 or 20 μM), PD146176 (10 or 20 μM) or BA (10 or 20 μM) treatment for 24 h. (f) BV2 cells were transfected with control (Ctrl) siRNA or 12/15-LO siRNA (si12/15-LO) and cultured for 24 h, then subjected to immunoblot analyses of 12/15-LO and PPARβ/δ. (g) BV2 cells were treated as described in e. Nuclear extracts were subjected to ELISA analyses of DNA-binding activities. Data are normalized to the activity of nuclear extracts from BV2 cells treated with vehicle. Results in c and g are shown as mean±S.E.M. (n=6). Results in a, b and d–f are representative of three independent experiments with similar results. **P<0.01
Inhibition of microglia activation by PPARβ/δ is associated with repression of NF-κB and MAPK activities
We then analyzed whether suppression of microglia activation was due to BA-induced PPARβ/δ in microglia. Exposure of primary microglia to LPS significantly decreased PPARβ/δ mRNA (Figure 6a). However, pre-treatment with BA led to a marked recovery of PPARβ/δ mRNA level in a dose-dependent manner (Figure 6a). By contrast, BA pre-treatment significantly reduced proinflammatory cytokine and chemokine production (Figure 3f). Thus, PPARβ/δ expression was negatively associated with production of proinflammatory factors in microglia, implying BA induces PPARβ/δ to suppress microglia activation. To test the hypothesis, siRNA against PPARβ/δ was transfected into BV2 cells (Figure 6b). The inhibitory effect of BA on LPS-induced chemokines such as CCL2, CCL3, CCL20 and CXCL10, and proinflammatory cytokines including TNF-α, IL-1β, IL-6 and IL-12p40 were abolished on PPARβ/δ knockdown (Figure 6b). These results show that PPARβ/δ induction in microglia via 12/15-LO inhibition was responsible for suppression of microglia activation, indicated by reduced proinflammatory factor expression by microglia, which in turn reduced CNS inflammation in EAE.

Inhibition of microglia activation by PPARβ/δ was associated with repression of NF-κB and MAPK activities. (a) Primary microglia were treated with vehicle or BA for 12 h. Cells were then stimulated with or without LPS (100 ng/ml) for 3 h. Levels of PPARβ/δ mRNA were analyzed by real-time PCR. Data are normalized to the gene expression in vehicle-treated microglia without LPS stimulation and shown as mean±S.E.M. (n=6). (b) Top, BV2 cells were transfected with control (Ctrl) siRNA or PPARβ/δ siRNA (siPPARβ/δ) and cultured for 24 h, then subjected to immunoblot analyses of PPARβ/δ. Middle and bottom, BV2 cells were transfected with control (Ctrl) siRNA or PPARβ/δ siRNA. Cells were pre-treated with vehicle or BA (20 μM) for 12 h, and then cultured in the absence or presence of LPS for 3 h. Expression of proinflammatory cytokines and chemokines were analyzed by real-time PCR. Data are normalized to the gene expression in vehicle-treated VB2 cells without LPS stimulation and shown as mean±S.E.M. (n=6). (c) BV2 cells were pre-treated with vehicle or BA (10 or 20 μM) for 12 h, and then cultured in the absence or presence of LPS for 10 or 30 min. Levels of IκBα, p-IκBα, p38, p-p38, JNK, p-JNK, ERK and p-ERK were determined by immunoblot analyses. Results in b (top) and c are representative of three independent experiments with similar results. *P<0.05; **P<0.01
To define molecular mechanisms by which BA-induced PPARβ/δ restricted the proinflammatory responses, we examined the activities of transcription factors, including NF-κB and activator protein 1, in microglia cell line BV2. These transcription factors are known downstream components of PPARβ/δ signaling and are critical for LPS-mediated gene induction. Activation of LPS-stimulated NF-κB was severely attenuated by BA treatment, as indicated by decreased phosphorylation and degradation of IκBα (Figure 6c). In addition, LPS-dependent phosphorylation of MAPKs, including ERK1/2, p38 and JNK, was also substantially inhibited by BA treatment (Figure 6c). These results suggest that the anti-inflammatory effect of BA-induced PPARβ/δ was mediated by repression of NF-κB and MAPK activities.
Discussion
In this study, we showed that inhibition of 12/15-LO by BA attenuated CNS inflammation of EAE through increased PPARβ/δ expression in microglia, which reduced microglia phagocytosis and production of proinflammatory cytokines and chemokines. This in turn impaired recruitment and reactivation of autoimmune cells in the CNS and ameliorated EAE.
EAE is considered as an autoimmune demyelinating disease of the CNS. Autoreactive T cells, such as Th1 and Th17 cells, are thought to have central roles in the disease process. However, the attenuated severity of EAE by BA was not a result of impaired autoimmune cell responses in the periphery. Administration of BA with both preventive and therapeutic protocols markedly improved the clinical symptoms of EAE with no delay of EAE onset, which suggests that BA does not affect early T-cell activation in the periphery. This hypothesis was further examined by ex vivo analyses of MOG reactivity of encephalomyeltic splenocytes and passive EAE studies. As the reactivation of autoimmune cells by CNS resident, antigen-presenting cell microglia and the recruitment of large numbers of inflammatory cells into the CNS are critical steps in the pathophysiology of EAE, we investigated the status of microglia under BA treatment. We found that BA was able to induce PPARβ/δ expression in microglia in EAE mice. The induction of PPARβ/δ in microglia suppressed microglia activation and reduced expression of chemokines CCL2, CCL3, CCL20 and CXCL10, which suppressed CNS inflammation. Induction of PPARβ/δ by BA was further confirmed in vitro in primary cultures of microglia and a microglia cell line, BV2. In addition, we showed that PPARβ/δ inhibition of microglia activation was associated with repression of NF-κB and MAPK activities. To our knowledge, the downregulation of PPARβ/δ by 12/15-LO metabolites in microglia is unprecedented in both physiological and pathological conditions, and our finding could provide a previously uncharacterized approach for treatment of CNS autoimmune diseases.
PPARs are ligand-dependent nuclear receptors that are critical regulators of development and homeostasis by modulating lipid metabolism.33 Accumulating evidence demonstrates that PPARs exhibit important roles in modulating inflammatory responses.10, 31, 34 They fine-tune expression of target inflammatory genes through either trans-activation or trans-repression activities. Among the three PPAR family members, the consequences of PPARβ/δ activation are less well understood. PPARβ/δ is widely distributed in most tissues with abundant expression in the CNS.35 Our data indicated that induction of PPARβ/δ in the CNS, more specifically in microglia, alleviated CNS inflammation of EAE. In contrast to the findings that agonists of PPARγ significantly reduce T-cell activation, proliferation and secretion of IFN-γ,36, 37, 38, 39, 40, 41 these results point to a distinct mechanism of action of PPARβ/δ in amelioration of EAE pathogenesis. Currently, clinical trials of several PPARγ agonists are in progress for MS and Alzheimer’s disease, and it is necessary to consider whether PPARβ/δ agonists offer advantages versus treatment with PPARγ ligands.
12/15-LO belongs to the lipoxygenase family of enzymes that promote polyunsaturated fatty acids to form a series of biologically active lipid mediators.9, 10 Based on their phylogenetic classification, murine leukocyte-type 12-LO and human reticulocyte-type 15-LO are highly conserved, sharing about 73% sequence similarity, and belong to the same 12/15-LO subfamily.42 Expression of murine 12/15-LO is limited to a few cell types such as macrophages, endothelial cells and adipocytes,43, 44, 45 but is highly expressed in macrophages.43, 44, 46 Notably, we found 12/15-LO was highly expressed in microglia, rather than astrocytes. Studies of 12/15-LO have been mainly focused on metabolism-related diseases such as diabetes47, 48 and atherosclerosis.46, 49 Recently, 12/15-LO was found to have important roles in regulation of allergic airway inflammation50 and CNS disorders such as Alzheimer’s disease17 and Parkinson’s disease.18 Here, we further extended the border of research to CNS autoimmune disease by using a pharmaceutical approach to selectively inhibit 12/15-LO by BA. This approach confirmed the pathogenic role of 12/15-LO in EAE, and demonstrated the feasibility of targeting 12/15-LO for treatment of inflammatory diseases in the CNS.
Interestingly, another lipoxygenase isoform, 5-LO, was known to be involved in the pathogenesis of Alzheimer’s disease and MS.51, 52 This study shows that BA only selectively inhibits the enzymatic activities of 12/15-LO but not those of 5-LO. These results, however, do not rule out the possibilities that 5-LO and its metabolites function in MS and EAE in a PPARβ/δ-independent manner.
Intriguingly, a previous study showed that 12/15-LO-deficient mice developed a late but severe clinical symptom as compared with wild-type mice.19 The authors detected a major reduction in axons in the cervical spinal cord, but found a very low level of T cells and no macrophages in the hindbrain of 12/15-LO-deficient mice during late active disease. They assumed that inactivation of PPARγ might be responsible for the exacerbation of EAE in 12/15-LO-deficient mice, but they did not provide further supporting evidence. In this study, we demonstrate that inhibition of 12/15-LO activity by a selective inhibitor, BA,23, 25 significantly ameliorate EAE in mice. Abundant PPARβ/δ but not PPARγ expression is induced in the CNS of EAE mice by BA treatment. Moreover, the main metabolite of 12/15-LO, 12-HETE, but not 5-LO metabolites, LTB4 or LTD4, could reverse BA-induced upregulation of PPARβ/δ in BV2 cells. Thus, our data seem to support that inhibition of 12/15-LO by a pharmacological approach could ameliorate EAE, and we elucidated a possible underlying mechanism.
Recently, it was reported that treatment of mice with a synthetic PPARβ/δ agonist, GW0742, ameliorated EAE severity.53 GW0742 did not prevent T-cell activation in the periphery, but it reduced glial cell activation. Another study showed that mice systemically deficient in PPARβ/δ developed severe EAE symptoms characterized by a striking accumulation of IFN-γ+CD4+ cells in the spinal cord, significantly higher proliferation and cytokine production of CD4+ T cells in the periphery, and enhanced expression of IL-12 family cytokines by myeloid cells.54 Together with our study, these results suggest that different modes of action by PPARβ/δ in different experimental conditions should be taken into consideration. As we mentioned previously, preferential induction of PPARβ/δ in CNS-resident microglia formed the underlying mechanism of treatment effects of BA in EAE. Consistently, BA is able to penetrate the blood–brain barrier to distribute in the CNS, and half-life of BA in the brain is about 125% longer than that in the blood,55 thus we assume that more BA crosses CNS to exert its pharmacological functions as blood–brain barrier is disrupted in the situation of EAE, which form the basis of protective properties of BA in CNS autoimmune diseases.
Among several pathways that have been identified for the anti-inflammatory actions of PPARβ/δ, inhibition of NF-κB is commonly involved. Inhibition of NF-κB activity by PPARβ/δ has been shown in endothelial cells,56 cardiomyocytes57 and adipocytes.58 Here, we demonstrated similar mechanisms in microglia, whereby BA-induced PPARβ/δ in microglia and was associated with repression of NF-κB and MAPK activities. Nevertheless, in addition to inhibition of proinflammatory transcription factors, whether PPARβ/δ induces anti-inflammatory genes in microglia needs to be further addressed. In this study, we have identified a previously uncharacterized pathway that inhibition of 12/15-LO induces PPARβ/δ in microglia, which alleviates EAE in mice. This reveals potential therapeutic applications for MS.
Materials and Methods
Induction and treatment of EAE
Female C57BL/6 mice (6–8 weeks of age) were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences (Shanghai, China), and were kept under specific pathogen-free conditions in the animal center of Shanghai Jiao Tong University School of Medicine (Shanghai, China). Age-matched mice were immunized s.c. with a synthetic peptide (300 μg) of MOG residues 35–55 (MEVGWYRSPFSRVVHLYRNGK) (GL Biochem, Shanghai, China). Immunization was performed by mixing the MOG peptide in CFA (Sigma, St. Louis, MO, USA) containing 5 mg/ml of Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI, USA). Pertussis toxin (200 ng; List Biological Laboratories, Campbell, CA, USA) in phosphate-buffered saline (PBS) was administered i.v. on days 0 and 2. For preventive or treatment protocols of EAE, BA (75 mg/kg; Sigma) or DMSO (as vehicle control) was administered i.p. daily from day 3 pre- or day 12 post-immunization onward. In some experiments, mice were immunized with the MOG peptide and treated with vehicle or BA daily from day 3 pre-immunization. On day 10 post immunization, DLN cells and splenocytes were isolated and re-challenged with the MOG (10 μg/ml) peptide for 3 days. Then, 5 × 106 viable cells were transferred into sublethally irradiated mice (6 Gy). The recipients received pertussis toxin immediately after cell transfer and 2 days later. For some experiments, DLN cells and splenocytes were isolated from EAE mice on day 10 post immunization. Cells were re-challenged with the MOG peptide and then transferred i.v. into sublethally irradiated mice. The recipients received pertussis toxin immediately after cell transfer and 2 days later. Recipients were also injected i.p. with vehicle or BA daily from day 7 post-transfer onward. Mice were examined daily and scored for disease severity by the standard scale: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with fore limb weakness or paralysis; 5, moribund or death. After the onset of EAE, food and water were provided on the cage floor. To eliminate any diagnostic bias, scores were assigned by researchers blinded to mouse identity. All animal procedures were approved by the Institutional Review Board of the Institute of Health Sciences.
Culture of primary microglia, astrocytes and BV2 cells
Cerebral cortical cells from newborn mice were dissociated by trypsinization and plated in complete DMEM. Two weeks later, culture flasks were shaken at 180–200 r.p.m. at 37 °C and loosely adherent cells were plated and incubated in complete DMEM at 37 °C for 1 h. After extensive washing, adherent microglia were collected. Tightly adherent astrocytes were also collected and incubated in complete DMEM. Purified microglia comprised a cell population in which 95% cells were CD11b-positive cells. Purified astrocytes comprised a cell population in which 95% cells were GFAP-positive cells. For further experiments, primary microglia, astrocytes, or BV2 cells were incubated in complete DMEM and stimulated with or without 100 ng/ml of LPS (Sigma) in the absence or presence of BA (10 or 20 μM), 12-HETE (100 pM), LTB4 (100 nM), LTD4 (100 nM), Zileuton (10 or 20 μM) (all from Sigma) or PD146176 (10 or 20 μM; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Histopathology and scoring
Paraffin-embedded transverse sections (8 μm) of spinal cords were stained with hematoxylin and eosin (H&E) or Luxol fast blue (LFB). Spinal cords stained with H&E were used for scoring inflammation (0: no inflammatory cells; 1: a few scattered inflammatory cells; 2: organization of inflammatory infiltrates around blood vessels; 3: extensive perivascular cuffing with extension into adjacent parenchyma, or parenchymal infiltration). Spinal cords stained with LFB were used for demyelination scoring (0: no demyelination; 1: a few scattered naked axons; 2: small groups of naked axons; 3: large groups of naked axons; 4: confluent foci of demyelination). Scores were generated by one well trained, board-certified histopathologist blind to the groups of mice.
Flow cytometry
Brains and spinal cords were collected from PBS-perfused mice, homogenized and filtered through a 100 μm cell strainer. Cells were collected by centrifugation. Cells were then resuspended in 37% Percoll (GE Amersham, Buckinghamshire, UK), overlayed onto 70% Percoll and then centrifuged. Cells at the 37–70% interface were collected and stained with antibodies (Abs) against mouse antigens such as CD4, CD8, CD45, CXCR3, CCR6 and CD11b (BD Pharmingen, San Diego, CA, USA). Stained cells were washed and analyzed with a FACSAria cytometer (BD Biosciences, San Diego, CA, USA).
Cell proliferation and measurement of cytokines and lipid metabolites
DLN cells and splenocytes isolated from vehicle- or BA-treated EAE mice were cultured in complete RPMI-1640 medium at a density of 5 × 105 per well in 96-well plates in the absence or presence of the MOG peptide at various concentrations. Cells were maintained at 37 °C in 5% CO2 for 72 h. Cell proliferation was measured with CCK8 reagent (Dojindo, Tokyo, Japan). For cytokine measurement, DLN cells and splenocytes isolated from vehicle- or BA-treated EAE mice were cultured in complete RPMI-1640 medium at a density of 1 × 106 per well in the presence of the MOG peptide (20 μg/ml). Supernatants were collected 48 h later and diluted for measurements of IFN-γ, IL-4, IL-10 and IL-17 by ELISA (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. For measurement of lipid metabolites, primary microglia, or BV2 cells were cultured in the absence or presence of 20 μM BA for 24 h, culture supernatants were then collected for detection of 12-HETE (Abcam, Cambridge, UK), 13-HODE (Abcam), 15-HETE (Mason Biotech, Houston, TX, USA), LTB4 (R&D Systems) and LTD4 (Mason Biotech).
Quantitative real-time PCR
Total RNA was isolated with TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions and reverse transcribed. Messenger RNA (mRNA) abundance was determined by real-time PCR with SYBR Green master mix (Applied Biosystems, Foster City, CA, USA) and specific primer sets (Table 1). Data were collected and quantitatively analyzed with an ABI Prism 7900 sequence detection system (Applied Biosystems). Gene expression was assessed by normalizing to β-actin. The control group was normalized to 1 in all figures, and data are presented as folds over control.
Electron microscopy
Mice were anaesthetized and perfused with 0.1 M of PBS followed by 0.1 M of PBS containing 2.5% glutaraldehyde. Lumbar spinal cords were processed, embedded and cut into 1-mm sections. Thin sections were sliced, stained with uranyl acetate and lead citrate, and analyzed by EM (CM120, Phillips, Eindhoven, The Netherlands) by one well trained, board-certified histopathologist blind to the groups of mice.
Immunoblotting
Total cell protein extracts were prepared and separated by 10% SDS-PAGE. Immunoblot analyses were performed by transfer of proteins onto PVDF membranes with a Trans-Blot apparatus (Bio-Rad, Hercules, CA, USA). After 1 h of blocking, membranes were incubated overnight at 4 °C with specific primary Abs with the following dilutions: anti-PPAR (Santa Cruz Biotechnology) and anti-PPARβ/δ Abs (Abcam) were used at 1 : 300; anti-12-lipoxygenase (Abcam), anti-5-LO, anti-phospho and total ERK1/2, p38 and JNK Abs (all from Cell Signaling Technology, Danvers, MA, USA), and anti-GAPDH (Cell Signaling Technology) Ab were used at 1 : 1000. Membranes were washed and subsequently incubated with appropriate HRP-conjugated secondary Abs for 1 h at room temperature, and washed again. Signals were detected with ECL reagents (Pierce Biotechnology, Rockford, IL, USA).
Immunofluorescence staining
Microglia were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked in 1% BSA, and incubated overnight with rat anti-mouse CD11b (BD Pharmingen) and PPARβ/δ Abs (Abcam). Subsequently, cells were incubated with fluorochrome-conjugated, goat anti-rat secondary Ab (Invitrogen) and counter stained with DAPI (Beyotime, Nantong, China). Images were taken by a confocal microscope (7CS SP5, Leica, Wetzlar, Germany) equipped with a 63 × oil immersion objective and analyzed by using LAS AF software (Leica) with an up to 3 × digital zoom. Minor adjustments to brightness were performed equally across the whole images. Fluorescence intensity was quantitated using ImageJ software (NIH, Bethesda, MD, USA).
Knockdown of 12/15-LO and PPARβ/δ genes by siRNA
The siRNAs to target murine 12/15-LO, PPARβ/δ or control siRNA were designed and synthesized (Genepharma, Shanghai, China). siRNA (100 nM) was transfected into the mouse microglia cell line BV2 with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
ELISA-based PPARβ/δ binding assay
Experiments were carried out with an ELISA kit (Cayman Chemicals, Ann Arbor, MI, USA) according to the manufacturer’s instructions. Briefly, nuclear extracts were prepared with NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific, Lafayette, CO, USA) according to the manufacturer’s instructions. Immunoblotting of GAPDH was used to detect cytoplasmic contamination in nuclear extracts. After that, 50 μg of nuclear extract proteins from vehicle control-, PD146176-, Zileuton- or BA-treated BV2 cells were added to peroxisome proliferator response element dsDNA-coated wells. Then, PPARβ/δ primary Ab and HRP-conjugated secondary Ab were sequentially added. The OD450 value was determined with an ELISA reader.
Statistical analyses
The Student’s t-test was used to analyze differences between groups. Where appropriate, one-way ANOVA was performed initially to determine whether an overall statistically significant change existed using a two-tailed paired or unpaired Student’s t-test. Non-parametric Mann–Whitney test was taken to analyze clinical score data between two groups. All tests were performed using SPSS version 13.0 (SPSS Inc., Chicago, IL, USA). A P-value <0.05 was considered statistically significant.
Abbreviations
- Ab:
-
antibody
- BA:
-
baicalein
- CNS:
-
central nervous system
- DLN:
-
draining lymph node
- EAE:
-
experimental autoimmune encephalomyelitis
- EM:
-
electron microscopy
- H&E:
-
hematoxylin and eosin
- HETE:
-
hydroxyeicosatetraenoic acid
- HODE:
-
hydroxyoctadecadienoic acid
- IFN:
-
interferon
- IL:
-
interleukin
- LFB:
-
Luxol fast blue
- LO:
-
lipoxygenase
- LTB4:
-
leukotriene B4
- LTD4:
-
leukotriene D4
- MOG:
-
myelin oligodendrocyte glycoprotein
- mRNA:
-
messenger RNA
- MS:
-
multiple sclerosis
- PBS:
-
phosphate-buffered saline
- PPAR:
-
peroxisome proliferator-activated receptor
- Th:
-
T helper
- TNF:
-
tumor necrosis factor
References
Weinstock-Guttman B, Zivadinov R, Mahfooz N, Carl E, Drake A, Schneider J et al. Serum lipid profiles are associated with disability and MRI outcomes in multiple sclerosis. J Neuroinflammation 2011; 8: 127.
Wheeler D, Bandaru VV, Calabresi PA, Nath A, Haughey NJ . A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain 2008; 131: 3092–3102.
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 2008; 172: 146–155.
Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2008; 105: 9041–9046.
Jack C, Ruffini F, Bar-Or A, Antel JP . Microglia and multiple sclerosis. J Neurosci Res 2005; 81: 363–373.
Maccarrone M, Dainese E, Oddi S . Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem Sci 2010; 35: 601–608.
Webb M, Luo L, Ma JY, Tham CS . Genetic deletion of fatty acid amide hydrolase results in improved long-term outcome in chronic autoimmune encephalitis. Neurosci Lett 2008; 439: 106–110.
Li B, Reynolds JM, Stout RD, Bernlohr DA, Suttles J . Regulation of Th17 differentiation by epidermal fatty acid-binding protein. J Immunol 2009; 182: 7625–7633.
Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL . Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res 2011; 50: 115–131.
Brash AR . Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem 1999; 274: 23679–23682.
Ferrucci L, Cherubini A, Bandinelli S, Bartali B, Corsi A, Lauretani F et al. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J Clin Endocrinol Metab 2006; 91: 439–446.
Pantaleo P, Marra F, Vizzutti F, Spadoni S, Ciabattoni G, Galli C et al. Effects of dietary supplementation with arachidonic acid on platelet and renal function in patients with cirrhosis. Clin Sci (Lond) 2004; 106: 27–34.
Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C et al. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999; 400: 378–382.
Nishiyama M, Watanabe T, Ueda N, Tsukamoto H, Watanabe K . Arachidonate 12-lipoxygenase is localized in neurons, glial cells, and endothelial cells of the canine brain. J Histochem Cytochem 1993; 41: 111–117.
Bendani MK, Palluy O, Cook-Moreau J, Beneytout JL, Rigaud M, Vallat JM . Localization of 12-lipoxygenase mRNA in cultured oligodendrocytes and astrocytes by in situ reverse transcriptase and polymerase chain reaction. Neurosci Lett 1995; 189: 159–162.
Chu J, Zhuo JM, Pratico D . Transcriptional regulation of beta-secretase-1 by 12/15-lipoxygenase results in enhanced amyloidogenesis and cognitive impairments. Ann Neurol 2012; 71: 57–67.
Yang H, Zhuo JM, Chu J, Chinnici C, Pratico D . Amelioration of the Alzheimer's disease phenotype by absence of 12/15-lipoxygenase. Biol Psychiatry 2010; 68: 922–929.
Li Y, Maher P, Schubert D . A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 1997; 19: 453–463.
Emerson MR, LeVine SM . Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res 2004; 1021: 140–145.
Kersten S, Desvergne B, Wahli W . Roles of PPARs in health and disease. Nature 2000; 405: 421–424.
Wahli W, Michalik L . PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab 2012; 23: 351–363.
Esposito E, Rinaldi B, Mazzon E, Donniacuo M, Impellizzeri D, Paterniti I et al. Anti-inflammatory effect of simvastatin in an experimental model of spinal cord trauma: involvement of PPAR-alpha. J Neuroinflammation 2012; 9: 81.
Gray E, Ginty M, Kemp K, Scolding N, Wilkins A . The PPAR-gamma agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J Neuroinflammation 2012; 9: 63.
Deschamps JD, Kenyon VA, Holman TR . Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg Med Chem 2006; 14: 4295–4301.
van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Baicalein LoEH . and 12/15-lipoxygenase in the ischemic brain. Stroke 2006; 37: 3014–3018.
Miller SD, Karpus WJ . The immunopathogenesis and regulation of T-cell-mediated demyelinating diseases. Immunol Today 1994; 15: 356–361.
van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation 2012; 9: 156.
Tian L, Ma L, Kaarela T, Li Z . Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J Neuroinflammation 2012; 9: 155.
Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 2005; 11: 146–152.
Xiao Y, Xu J, Wang S, Mao C, Jin M, Ning G et al. Genetic ablation of steroid receptor coactivator-3 promotes PPAR-beta-mediated alternative activation of microglia in experimental autoimmune encephalomyelitis. Glia 2010; 58: 932–942.
Yang Y, Lovett-Racke AE, Racke MK . Regulation of immune responses and autoimmune encephalomyelitis by PPARs. PPAR Res 2010; 2010: 104705.
Xu YW, Sun L, Liang H, Sun GM, Cheng Y . 12/15-Lipoxygenase inhibitor baicalein suppresses PPAR gamma expression and nuclear translocation induced by cerebral ischemia/reperfusion. Brain Res 2010; 1307: 149–157.
Poulsen L, Siersbaek M, Mandrup S . PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol 2012; 23: 631–639.
Defaux A, Zurich MG, Braissant O, Honegger P, Monnet-Tschudi F . Effects of the PPAR-beta agonist GW501516 in an in vitro model of brain inflammation and antibody-induced demyelination. J Neuroinflammation 2009; 6: 15.
Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W . Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996; 137: 354–366.
Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ . The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol 2000; 164: 1364–1371.
Marx N, Mach F, Sauty A, Leung JH, Sarafi MN, Ransohoff RM et al. Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J Immunol 2000; 164: 6503–6508.
Natarajan C, Bright JJ . Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun 2002; 3: 59–70.
Schmidt S, Moric E, Schmidt M, Sastre M, Feinstein DL, Heneka MT . Anti-inflammatory and antiproliferative actions of PPAR-gamma agonists on T lymphocytes derived from MS patients. J Leukoc Biol 2004; 75: 478–485.
Harris SG, Phipps RP . Peroxisome proliferator-activated receptor gamma (PPAR-gamma) activation in naive mouse T cells induces cell death. Ann N Y Acad Sci 2000; 905: 297–300.
Tautenhahn A, Brune B, von Knethen A . Activation-induced PPARgamma expression sensitizes primary human T cells toward apoptosis. J Leukoc Biol 2003; 73: 665–672.
Kuhn H, Thiele BJ . The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett 1999; 449: 7–11.
Sun D, Funk CD . Disruption of 12/15-lipoxygenase expression in peritoneal macrophages. Enhanced utilization of the 5-lipoxygenase pathway and diminished oxidation of low density lipoprotein. J Biol Chem 1996; 271: 24055–24062.
Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation 2004; 110: 2024–2031.
Bolick DT, Orr AW, Whetzel A, Srinivasan S, Hatley ME, Schwartz MA et al. 12/15-Lipoxygenase regulates intercellular adhesion molecule-1 expression and monocyte adhesion to endothelium through activation of RhoA and nuclear factor-kappaB. Arterioscler Thromb Vasc Biol 2005; 25: 2301–2307.
Poeckel D, Zemski Berry KA, Murphy RC, Funk CD . Dual 12/15- and 5-lipoxygenase deficiency in macrophages alters arachidonic acid metabolism and attenuates peritonitis and atherosclerosis in ApoE knock-out mice. J Biol Chem 2009; 284: 21077–21089.
Tokuyama Y, Sturis J, DePaoli AM, Takeda J, Stoffel M, Tang J et al. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes 1995; 44: 1447–1457.
Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL . Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J Clin Invest 1999; 103: 1431–1436.
Rong S, Cao Q, Liu M, Seo J, Jia L, Boudyguina E et al. Macrophage 12/15 lipoxygenase expression increases plasma and hepatic lipid levels and exacerbates atherosclerosis. J Lipid Res 2012; 53: 686–695.
Andersson CK, Claesson HE, Rydell-Tormanen K, Swedmark S, Hallgren A, Erjefalt JS . Mice lacking 12/15-lipoxygenase have attenuated airway allergic inflammation and remodeling. Am J Respir Cell Mol Biol 2008; 39: 648–656.
Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Pratico D . 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer's disease. FASEB J 2008; 22: 1169–1178.
Mirshafiey A, Jadidi-Niaragh F . Immunopharmacological role of the leukotriene receptor antagonists and inhibitors of leukotrienes generating enzymes in multiple sclerosis. Immunopharmacol Immunotoxicol 2010; 32: 219–227.
Polak PE, Kalinin S, Dello Russo C, Gavrilyuk V, Sharp A, Peters JM et al. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J Neuroimmunology 2005; 168: 65–75.
Dunn SE, Bhat R, Straus DS, Sobel RA, Axtell R, Johnson A et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J Exp Med 2010; 207: 1599–1608.
Tsai TH, Liu SC, Tsai PL, Ho LK, Shum AY, Chen CF . The effects of the cyclosporin A, a P-glycoprotein inhibitor, on the pharmacokinetics of baicalein in the rat: a microdialysis study. Br J Pharmacol 2002; 137: 1314–1320.
Rival Y, Beneteau N, Taillandier T, Pezet M, Dupont-Passelaigue E, Patoiseau JF et al. PPARalpha and PPARdelta activators inhibit cytokine-induced nuclear translocation of NF-kappaB and expression of VCAM-1 in EAhy926 endothelial cells. Eur J Pharmacol 2002; 435: 143–151.
Smeets PJ, Teunissen BE, Planavila A, de Vogel-van den Bosch H, Willemsen PH, van der Vusse GJ et al. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J Biol Chem 2008; 283: 29109–29118.
Rodriguez-Calvo R, Serrano L, Coll T, Moullan N, Sanchez RM, Merlos M et al. Activation of peroxisome proliferator-activated receptor beta/delta inhibits lipopolysaccharide-induced cytokine production in adipocytes by lowering nuclear factor-kappaB activity via extracellular signal-related kinase 1/2. Diabetes 2008; 57: 2149–2157.
Acknowledgements
We thank Professor Gary Brewer (Robert Wood Johnson Medical School, NJ) for critical reading of the manuscript. We thank Drs. Ying Wang, Congfeng Xu, Haikuo Xue, Hong Zhou, and instructors Laiwen Fu, Xiang Miao, and Shuyang Yan (Institute of Health Sciences, Shanghai, China) for their assistance in experiments. This work was supported by grants from the Ministry of Science and Technology of China (2011CB966200), Strategic Priority Research Program of the Chinese Academy of Science (XDA01040000), the National Science Foundation of China (81130057, 30901317 and 81071748), the Program of Science and Technology Commission of Shanghai Municipality (11JC1411400, 11431920900 and 08JC1413300), the Program of Chinese Academy of Science (KSCX2-YW-R-245 and KSCX2-YW-R-175) and the Shanghai Leading Academic Discipline Project (J50207).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Finazzi-Agró
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Xu, J., Zhang, Y., Xiao, Y. et al. Inhibition of 12/15-lipoxygenase by baicalein induces microglia PPARβ/δ: a potential therapeutic role for CNS autoimmune disease. Cell Death Dis 4, e569 (2013). https://doi.org/10.1038/cddis.2013.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.86
Keywords
This article is cited by
-
Lipoxygenase Metabolism: Critical Pathways in Microglia-mediated Neuroinflammation and Neurodevelopmental Disorders
Neurochemical Research (2022)
-
Retinoid X Receptor: Cellular and Biochemical Roles of Nuclear Receptor with a Focus on Neuropathological Involvement
Molecular Neurobiology (2022)
-
Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain
Nature Communications (2020)
-
Baicalein ameliorates pristane-induced lupus nephritis via activating Nrf2/HO-1 in myeloid-derived suppressor cells
Arthritis Research & Therapy (2019)
-
Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis
Cell Death & Disease (2019)
