
Abstract
We show that cyclic AMP (cAMP) elevating agents protect blasts from patients with acute promyelocytic leukemia (APL) against death induced by first-line anti-leukemic anthracyclines like daunorubicin (DNR). The cAMP effect was reproduced in NB4 APL cells, and shown to depend on activation of the generally cytoplasmic cAMP-kinase type I (PKA-I) rather than the perinuclear PKA-II. The protection of both NB4 cells and APL blasts was associated with (inactivating) phosphorylation of PKA site Ser118 of pro-apoptotic Bad and (activating) phosphorylation of PKA site Ser133 of the AML oncogene CREB. Either event would be expected to protect broadly against cell death, and we found cAMP elevation to protect also against 2-deoxyglucose, rotenone, proteasome inhibitor and a BH3-only mimetic. The in vitro findings were mirrored by the findings in NSG mice with orthotopic NB4 cell leukemia. The mice showed more rapid disease progression when given cAMP-increasing agents (prostaglandin E2 analog and theophylline), both with and without DNR chemotherapy. The all-trans retinoic acid (ATRA)-induced terminal APL cell differentiation is a cornerstone in current APL treatment and is enhanced by cAMP. We show also that ATRA-resistant APL cells, believed to be responsible for treatment failure with current ATRA-based treatment protocols, were protected by cAMP against death. This suggests that the beneficial pro-differentiating and non-beneficial pro-survival APL cell effects of cAMP should be weighed against each other. The results suggest also general awareness toward drugs that can affect bone marrow cAMP levels in leukemia patients.
Similar content being viewed by others


Main
Cyclic AMP (cAMP) is the second messenger for extracellular signals, such as endogenous and exogenous β-adrenergic stimulators and members of the prostaglandin family, often increased under conditions of inflammation. The cellular cAMP concentration can also be increased by phosphodiesterase (PDE)-inhibiting drugs. The major cAMP mediators are the cAMP-dependent protein kinase isozyme I and II (PKA-I, PKA-II), and the cAMP-stimulated GDP exchange factor Epac.1, 2
A striking effect of cAMP3 and the cAMP synthesis-stimulating prostaglandin E2 (PGE2)4 is enhanced all-trans retinoic acid (ATRA)-induced maturation of acute promyelocytic leukemia (APL)-derived NB4 cells.5 ATRA-induced maturation is a cornerstone in APL therapy, and its combination with cAMP signaling stimulators has been advocated to improve current APL therapy. Thus, stimulation of cAMP signaling by PDE inhibitor enhanced the effect of ATRA on survival of syngenic PML-RARA APL mice and mice transplanted with NB4 cells,6, 7, 8 and retarded the APL progression in a patient.7 Although cAMP stimulation protects mature neutrophils9, 10, 11 and promonocytic leukemia cells12 against death and induces death of the BNML-derived AML line IPC,13 little is known about the impact of cAMP on APL cell survival. This is of particular concern as ATRA is used together with an anthracycline (daunorubicin; DNR or idarubicin; IDA) in current APL treatment protocols.14, 15
Here we used the in vivo APL model of NB4 cells transplanted into NOD-scid IL2rγnull (NSG) mice16 to find the impact of cAMP-elevating agents on APL progression in the absence and presence of DNR treatment. Animals injected with stable PGE2 analog and cAMP phosphodiesterase inhibitor had shortened life span both in the absence and presence of DNR treatment. The in vitro studies showed that the cAMP agonists protected NB4 cells against a number of death-inducing cell stressors, including first-line anthracycline drugs like DNR. The protection was mediated by activation of cAMP-dependent protein kinase type I (PKA-I), and accompanied by inactivating phosphorylation of the pro-apoptotic protein Bad and activating phosphorylation of the AML proto-oncogene CREB, both on known PKA-targeted residues. The clinical relevance of the NB4 model is supported by studies of blasts from APL and AML patients, which also were protected by cAMP against DNR-induced death in vitro. The findings indicate awareness when patients on cAMP-elevating agents like theophylline are treated with anthracyclines, or when cAMP stimulation is considered combined with ATRA to boost APL cell differentiation.
Results
cAMP agonists protect NB4 APL cells against DNR-induced death
We found that human NB4 cells, widely accepted as being representative of human APL,5 were strongly protected by cAMP agonists against death induced by the first-line anti-leukemic anthracycline drug DNR. The degree of protection (Figure 1a, main panel) correlated with the cellular cAMP level, which was raised more when the adenylate cyclase stimulator PGE2 was combined with the cAMP phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; Figure 1a, upper right inset). The potentiating effect of IBMX and the efficacy of the cAMP analog N6-Bz-cAMP (Figure 1a, main panel) both indicated that the protective effect of PGE2 was mediated by cAMP. As a prelude to intact animal studies, we found that the stable PGE2 analog 16,16-dimethyl PGE2 (dmPGE2) synergized with any of four different cAMP phoshodiesterase inhibitors, including the clinically relevant compounds theophylline (aminophylline), milrinone and rolipram (Figure 1b).

Cyclic AMP agonists protect NB4 cells against DNR-induced cell death in proportion to the intracellular cAMP elevation, and do not interfere with DNR uptake. (a) NB4 cells were treated for 3, 4 or 5 h with  : vehicle,
: vehicle,  : DNR (5 μM),
: DNR (5 μM),  : DNR+PGE2 (1.5 μM, added 0.5 h before DNR),
: DNR+PGE2 (1.5 μM, added 0.5 h before DNR),  : DNR+PGE2+IBMX (0.3 mM, added 0.5 h before DNR) or
: DNR+PGE2+IBMX (0.3 mM, added 0.5 h before DNR) or  : DNR+N6-Bz-cAMP (0.4 mM, added 0.5 h before DNR). Cell death was scored by microscopy (the inserted photomicrographs show cells treated for 5 h), and is given as mean±S.E.M., n=7. Significance by Student t-test: **P<0.01, *P<0.05, n.s.=not significant. PGE2, IBMX or N6-Bz-cAMP alone had no effect on % dead cells (data not shown). The upper right hand inset shows the cAMP level in cells treated for 1 h under some of the conditions above (
: DNR+N6-Bz-cAMP (0.4 mM, added 0.5 h before DNR). Cell death was scored by microscopy (the inserted photomicrographs show cells treated for 5 h), and is given as mean±S.E.M., n=7. Significance by Student t-test: **P<0.01, *P<0.05, n.s.=not significant. PGE2, IBMX or N6-Bz-cAMP alone had no effect on % dead cells (data not shown). The upper right hand inset shows the cAMP level in cells treated for 1 h under some of the conditions above ( ,
,  ,
,  ,
,  ), the bars representing the range of two experiments. (b) The cells were treated with DNR (5 μM, 5 h) in the presence or absence of dmPGE2 (1 μM) together with the PDE inhibitors IBMX (0.1 mM), theophylline (‘Aminophylline’, 0.15 mM), milrinone (0.15 mM) or rolipram (0.15 mM) as indicated, and cell death scored by microscopy. DmPGE2, IBMX, aminophylline, milrinone or rolipram did not induce death alone (data not shown). The data are given as mean±S.E.M., n=3. Significance by Student t-test: **P<0.01, *P<0.05. (c) FACS forward and side scatter analysis for size and granularity of NB4 (upper 3 panels) and NB4-LR1 (middle 3 panels) cells treated for 4 h with vehicle (Ctrl) or DNR (5 μM) in the absence or presence of N6-Bz-cAMP (0.4 mM). The lower 3 panels show a similar analysis for NB4-LR2 cells treated for 4 h with vehicle (Ctrl) or DNR (5 μM) in the absence or presence of PGE2/IBMX (2 μM/0.1 mM). The data shown are representative for four separate experiments. (d) NB4 cells were treated for 4.5 h with vehicle (Ctrl) or N6-Bz-cAMP (0.4 mM), or with DNR (5 μM) in the absence or presence of N6-Bz-cAMP (0.4 mM) or PGE2/IBMX (2 μM/0.1 mM), and analyzed for Annexin V binding by FACS. Necrotic cells with positive PI staining (<5%) were omitted from the analyses. A representative experiment of three is shown. (e) NB4 cells were treated for 3 or 6 h with DNR (5 μM) alone or in combination with N6-Bz-cAMP (0.4 mM), and cell extracts analyzed by immunoblotting for Pro-caspase 3 and Hsp90 cochaperone p23. Actin was used as loading control. The position of selected MW standard is indicated. (f) The strong fluorescence of DNR was used to determine its concentration by flow cytometry of NB4 cells incubated for 2 or 5 h with DNR (5 μM) in the presence or absence of N6-Bz-cAMP (0.4 mM). Note the lack of effect of N6-Bz-cAMP on DNR content
), the bars representing the range of two experiments. (b) The cells were treated with DNR (5 μM, 5 h) in the presence or absence of dmPGE2 (1 μM) together with the PDE inhibitors IBMX (0.1 mM), theophylline (‘Aminophylline’, 0.15 mM), milrinone (0.15 mM) or rolipram (0.15 mM) as indicated, and cell death scored by microscopy. DmPGE2, IBMX, aminophylline, milrinone or rolipram did not induce death alone (data not shown). The data are given as mean±S.E.M., n=3. Significance by Student t-test: **P<0.01, *P<0.05. (c) FACS forward and side scatter analysis for size and granularity of NB4 (upper 3 panels) and NB4-LR1 (middle 3 panels) cells treated for 4 h with vehicle (Ctrl) or DNR (5 μM) in the absence or presence of N6-Bz-cAMP (0.4 mM). The lower 3 panels show a similar analysis for NB4-LR2 cells treated for 4 h with vehicle (Ctrl) or DNR (5 μM) in the absence or presence of PGE2/IBMX (2 μM/0.1 mM). The data shown are representative for four separate experiments. (d) NB4 cells were treated for 4.5 h with vehicle (Ctrl) or N6-Bz-cAMP (0.4 mM), or with DNR (5 μM) in the absence or presence of N6-Bz-cAMP (0.4 mM) or PGE2/IBMX (2 μM/0.1 mM), and analyzed for Annexin V binding by FACS. Necrotic cells with positive PI staining (<5%) were omitted from the analyses. A representative experiment of three is shown. (e) NB4 cells were treated for 3 or 6 h with DNR (5 μM) alone or in combination with N6-Bz-cAMP (0.4 mM), and cell extracts analyzed by immunoblotting for Pro-caspase 3 and Hsp90 cochaperone p23. Actin was used as loading control. The position of selected MW standard is indicated. (f) The strong fluorescence of DNR was used to determine its concentration by flow cytometry of NB4 cells incubated for 2 or 5 h with DNR (5 μM) in the presence or absence of N6-Bz-cAMP (0.4 mM). Note the lack of effect of N6-Bz-cAMP on DNR content
The effects could not be explained by coincidental blocking of one or a few features of cell death, as all the death phenotypes probed for were inhibited by cAMP, including cell membrane blebbing (Figures 1a and b), cell size reduction with diminished forward light scattering (Figure 1c) and phosphatidylserine externalization (Figure 1d). cAMP stimulation also prevented DNR-induced cleavage of pro-caspase 3 and the caspase target Hsp90 co-chaperone p23, known to have a role in leukemic apoptosis13, 17 (Figure 1e). In view of reports that cAMP can affect drug transporters18, 19 and concern that cAMP might lead to intracellular sequestration of DNR in intracellular vacuoles, we determined the total cell and the nuclear DNR content. We found identical total DNR concentration in cells incubated with DNR both in the absence and presence of N6-Bz-cAMP (Figure 1f), as well as similar intracellular distribution of DNR, with strong nuclear accumulation and even cytoplasmic content (data not shown).
cAMP failed to protect the AML cell lines Mv4-11 and HL60 against DNR death, whether based on morphological assessment (Figures 2a and b) or (for HL60) cleavage of p23 (Figure 2c). As cAMP agonists enhance DNR-induced cell death in the BNML-derived rat IPC-81 cell AML model,13 we conclude that AML cell lines differ greatly in their response to the combination of DNR and cAMP stimulation.

Cyclic AMP agonists can protect APL and AML blasts against anthracycline-induced death. (a and b) Mv4-11 (a) and HL60 (b) AML cells were treated for 4 or 7 h, respectively, with DNR in the absence or presence of PGE2/IBMX (2 μM/0.15 mM) or N6-Bz-cAMP (0.4 mM). The % dead cells was scored by microscopy (chromatin condensation), and is given as mean±S.E.M., n=3. Significance by Student t-test: **P<0.01. (c) Extracts isolated from HL60 cells treated with DNR (5 μM) for 3 or 5 h alone or in combination with PGE2/IBMX (2 μM/0.15 mM) were analyzed by immunoblotting for Hsp90 cochaperone p23. Actin was used as loading control. (d–m) Blasts were isolated from peripheral blood of three patients with diagnosed APL (APL1-APL3) and seven AML patients (AML1-AML7) and incubated with DNR alone or together with PGE2/IBMX (2 μM/0.3 mM) for 3 h (APL1, APL2 (8 μM DNR)) or 24 h (APL2 (0.2 and 0.8 μM DNR), APL3; AML 1–7). Samples were assessed for drug-induced cell death by FACS analysis of PI uptake (APL1, APL 2) or by Annexin V labeling and PI uptake (APL2, APL3, AML1-AML-7). When present, PGE2/IBMX was added 0.5 h before DNR. Further experimental details are given in the Materials and Methods section. Similar results were obtained when PGE2/IBMX was replaced by the cAMP analog N6-Bz-cAMP (data not shown). A Wilcoxon paired two-tailed comparison test of the ratio of dead cells after incubation with DNR alone and DNR+either PGE2/IBMX or N6-Bz-cAMP (0.4 mM) for all the experiments performed on the APL1-3 blasts, yielded P<0.004 for a protective effect of the cAMP agonists against P<0.007 for the AML1-7 blasts
cAMP agonists can protect APL and AML patient blasts against DNR-induced death
To probe which, if any, of the studied cell line models is relevant for human acute myeloid leukemia, we tested leukemic blasts isolated from patients with diagnosed APL or AML (see Supplementary Table S1 for details of disease). None of the blast isolates showed cAMP-enhanced DNR-induced death, as observed in the IPC cells. Rather, all the three APL blast isolates (Figures 2d–f) and six of the seven AML blast isolates (Figures 2g–m) resembled the NB4 cells by being protected against DNR by PGE2/IBMX. The APL blasts had an average of 47±5.9% (mean±S.E.M.; n=3) protection and the AML blasts 41±5.8% (n=7). With N6-Bz-cAMP, the average protection of APL blasts was 51% (data not shown). The NB4 cells appear therefore to be the most relevant cell line to study cAMP effects on anthracycline-induced acute myeloid leukemic blast death.
cAMP agonists protect NB4 cells under in vitro conditions relevant for the leukemic bone marrow and enhance APL progression in a NB4 orthotropic NSG model
In order to better judge the intact organism relevance, additional in vitro experiments were conducted to cover DNR and IDA concentrations likely to be encountered in vivo20 and to mimic the hypoxic nature of leukemic cell bone marrow niches.21 We demonstrated that cAMP agonists protected NB4 cells also under hypoxic conditions, and against a wide range of anthracycline concentrations (0.1–5 μM DNR and 10–500 nM IDA), and for periods up to 32 h (Figure 3a, Supplementary Figure S1a).

Cyclic AMP-elevating agents protect NB4 cells against death induced by a number of cell stressors under conditions relevant for the leukemic bone marrow. (a) NB4 cells were treated for various periods of time and various concentrations of DNR in the absence or presence of PGE2/IBMX (2 μM/0.15 mM) in standard (19% O2, 5% CO2; solid bars) or hypoxic (2% O2, 5% CO2; hatched bars) atmosphere. The % dead cells was scored by microscopy (chromatin condensation and surface morphology), and is given as mean±S.E.M., n=3. Significance by Student t-test: **P<0.01, *P<0.05. (b) NB4 and NB4-LR2 cells were primed with ATRA (1 μM, 24 h). They were next incubated for 5 h with DNR (D; 5 μM) or 6 h with IDA (I; 0.5 μM) or vehicle in the absence or presence of PGE2/IBMX (2 μM/0.1 mM). The % dead cells are shown as upwards solid bars. The % of maturated cells was determined by the nitroblue tetrazolium (NBT) reduction assay, and is given as open, downward bars. The ATRA-resistant NB4-LR2 cells were, as expected, NBT negative. The data are given as mean±S.E.M., n=3. Significance by Student t-test: **P<0.01, *P<0.05. (c) NB4 cells were treated with the HSP90 antagonist geldanamycin (5 μM), the mitochondrial toxin rotenone (5 μM), the proteasome inhibitor bortezomib (0.2 μM) or the glucose antagonist 2-deoxyglucose (10 mM) for the periods of time indicated in the absence or presence of dmPGE2 (2 μM)/IBMX (0.15 mM) or dmPGE2 (2 μM)/aminophylline (0.15 mM). The scoring of dead cells was as a and b
In current standard protocols for APL treatment, DNR or IDA is administered shortly after the onset of ATRA treatment, and later used jointly with ATRA to eliminate ATRA-resistant cells.15 We tested, therefore, whether cAMP agonists could counteract the DNR-induced death both of NB4 cells starting to undergo differentiation with ATRA (Figure 3b, left panel), and of the ATRA-resistant sub-clones NB4-LR2 and NB4-LR15 (Figures 1c and 3b, right panel). We found that cAMP stimulation may protect both ATRA-primed NB4 cells and ATRA-resistant NB4 cells against DNR in the presence of ATRA (Figure 3b).
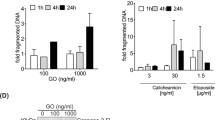
To know whether the cAMP agonists protected specifically against DNR, we tested whether they also could protect against agents promoting unfolded protein stress, like the proteasome inhibitor bortezomib and the HSP90 inhibitor geldanamycin, and strong metabolic stressors like 2-deoxyglucose and rotenone. We found significant protection by cAMP against these stressors both for NB4 cells (Figure 3c) and APL and AML blasts (Supplementary Figure S1b). In conclusion, the elevation of cAMP is expected to promote NB4 cell (and APL blast) survival under conditions thought to prevail in the leukemic bone marrow both without and with DNR therapy.
We tested, therefore, whether cAMP agonists could promote APL cell survival also in vivo, under the assumedly adverse conditions existing in the bone marrow under APL invasion and the even more adverse conditions under DNR treatment. For this purpose, we transplanted NOD-scid IL2rγnull mice (NSG) mice with NB4 cells and injected them with vehicle (control) or dmPGE2/theophylline. The NB4 cell-injected animals given only vehicle survived from 31–33 days (Figure 4a). The death was preceded by weight loss (Figure 4b). The animals were watched for development of extreme fatigue and/or dorsal limb paralysis before euthanization. The animals injected with cAMP agonists had shorter life span and more rapid weight loss than the vehicle-injected animals (Figures 4a and b). This difference was attributed to more rapid APL disease development, as the timing of fatigue and paralysis relative to death was similar, and the animals selected for autopsy showed similarly swollen bone marrow with brittle femurs and splenomegaly (data not shown).

cAMP enhances APL progression in an NB4 orthotropic NSG model. (a) Survival of NB4-transplanted NOD-scid IL2rγnull mice (NSG) treated with vehicle (‘Ctrl’, n=5), dmPGE2/aminophylline (125 μg/kg/20 mg/kg, ‘cAMP’, n=6), daunorubicin (2.5 mg/kg, ‘DNR’, n=6) or daunorubicin+dmPGE2/aminophylline (‘DNR+cAMP’, n=6). The group that received cAMP agonists had significantly (P=0.003) shorter life span compared with the Ctrl group (average 32.8 against 28.8 days). DNR increased the average life span compared with the Ctrl group from 32.8 to 45.7 days, against 28.8 to 38.8 days for the DNR+cAMP group. (b) The weight of the NB4-transplanted NSG mice described in a relative to that (%) at the time of NB4 cell injection. The data show the average for all mice in each group
The orthotopic NSG NB4 cell APL model was also used to test whether cAMP agonists could interfere with the effect of a 3 × 2.5 mg/kg DNR treatment regime, designed to resemble that used clinically in AML.14, 22 The DNR treatment had a strong survival-prolonging effect, which was significantly shortened for animals also receiving cAMP agonists (Figures 4a and b). In conclusion, cAMP stimulation protects NB4 cells against adverse in vivo conditions likely to be encountered in the leukemic bone marrow. It also accelerates the development of leukemia from injected NB4 cells in the intact NSG mouse, both without and with DNR therapy.
cAMP can counteract DNR-induced NB4 cell death via activation of PKA-I
cAMP has three major intracellular receptors, the cAMP-binding small G protein exchange factor Epac and the regulatory subunit of PKA-I (RIα,β) and PKA-II (RIIα,β). We excluded Epac as mediator, as the Epac-selective agonist23 8-CPT-2′-Me-cAMP was inefficient (data not shown), whereas the PKA-selective N6-Bz-cAMP23 protected NB4 cells as efficiently as increased endogenous cAMP (Figure 1).
The NB4 cell expresses both RIα and RIIα,β (Figure 5a, left lane), with RI diffusely speckled in the cytoplasm (Figure 5b, left panel) and RII confined to the perinuclear region (Figure 5c, left panel). The defined localization of the RII subunits, and thereby PKA-II, can channel PKA-II to a subset of substrates,24, 25 which might be essential for cAMP protection against death. In order to probe whether PKA-II had a specific role, we produced NB4RIIα,βKD cells by RNA-mediated double knockdown of RIIα and RIIβ.

PKA isoenzyme II is dispensable for cAMP-mediated protection against DNR-induced NB4 cell death. (a) Immunoblot analysis showing the effect of knockdown of the RIIα and RIIβ subunit of PKA in NB4RIIKD cells, as compared with NB4 cells transduced with empty vector (NB4V0 cells). Note that the decline of RII subunits in the NB4RIIKD cells is compensated by induction of RIα in the NB4RIIKD cells. (b and c) Immunofluorescence analysis showing the sub-cellular localization of the RI (b) and RII (c) subunits of PKA in NB4V0 and NB4RIIKD cells. Note the peri-nuclear position of the RII subunits. DAPI is used to visualize nuclear chromatin. (d) NB4V0 and NB4RIIKD cells were treated with DNR (5 μM, 5 h) alone or with N6-Bz-cAMP (0.4 mM), and cell death was scored. The data are given as mean±S.E.M., n=3
The NB4RIIα,βKD cells lacked nearly all the RIIα and most of the RIIβ (Figures 5a and c, right panels), but had a compensatory increase of RIα (Figure 5a, right panel) and unchanged total PKA concentration and basal PKA activity (data not shown). The knockdown of RIα was not undertaken, in part because a selective inhibitor Rp-8-Br-cAMPS26 is available, and in part because downregulation of RIα in our hands is not compensated by RII upregulation. Hence, the resulting excess of free cAMP-unregulated C subunit leads to high PKA activity already in the absence of cAMP stimulation.
The NB4RIIα,βKD cells were as well protected against DNR by N6-Bz-cAMP as the NB4wt (Figure 1) and control-transfected NB4V0 cells (Figure 5d), indicating that activation of PKA-I alone suffices to protect. This does not exclude that both isozymes can contribute in the NB4wt or NB4V0 cells. We probed therefore such cells for cAMP analog pair synergy regarding survival, using both PKA-I- and PKA-II-directed pairs. The isozyme-specific synergy of activation is based on their differential complementarity for binding to sites AI and BI of PKA-I or AII and BII of PKA-II of each member of a pair (see Supplementary Tables S2 and S3 for details of the analogs, including the new 2-Cl-8-HA-cAMP). The PKA-I directed analog pair synergized both in the NB4V0 and NB4RIIα,βKD cells, the length of the horizontal arrows indicating the magnitude of the analog complementarity (Figures 6a and b; see Supplementary Figure S2a,b for alternative plots and additional data). In contrast, the PKA-II-directed analog pair failed to show synergism (Figure 6c, Supplementary Figure S2c). We conclude therefore that RI subunit of PKA-I is the cAMP receptor mediating the protective action of cAMP in NB4 cells. The conclusion is supported by the ability of the PKA-I-specific competitive inhibitor Rp-8-Br-cAMPS26 to counteract the protective effect of N6-Bz-cAMP against DNR (Figure 6d). The pivotal role of PKA suggested that the protective action of cAMP was mediated primarily by phosphorylation of PKA substrate(s).

The relative role of PKA isoenzyme I and II to protect against DNR-induced NB4 cell death. (a and b) NB4 cells with intact PKA isozymes (NB4V0; a) or with knocked-down RII subunits (NB4RIIKD; b) were incubated for 4 h with DNR (5 μM) and various concentrations of 2-Cl-8-HA-cAMP in the absence (•, ) or presence (○,▵) of N6-Bz-8-PIP-cAMP (70 μM), which cooperates with 2-Cl-8-HA-cAMP to activate PKA-I, but not PKA-II. The percentage of dead cells dropped more rapidly when 2-Cl-8-HA-cAMP was combined with N6-Bz-8-PIP-cAMP (horizontal arrows), indicating that selective activation of PKA-I could protect both NB4V0 and NB4RIIKD cells against DNR. To better assess the protective synergy between the two analogs (horizontal arrow) the initial cell death was set to 100%. The data represent the average of two experiments, and the original data are shown in Supplementary Figure S2a,b. Similar results were obtained with other PKA-I synergizing analog combinations (data not shown). The enhanced protection with N6-Bz-8-PIP-cAMP was judged by Wilcoxon paired comparison test for the 2-Cl-8-HA-cAMP concentration range 30–600 μM, and was found significant (P<0.01) both for the NB4V0 and the NB4RIIKD cells. (c) NB4V0 cells were treated as described in a, except that the concentration of Sp-5,6-DCl-cBIMPS was varied in the absence (●) or presence (○) of N6-MBC-cAMP (8.5 μM), in order to test for PKA-II synergism. The data represent the average of two experiments (Supplementary Figure S2c). Similar results (no synergy) were obtained with other PKA-II synergizing analog combinations (data not shown). (d) NB4WT cells were treated for 4 h with DNR (5 μM) and increasing concentrations of N6-Bz-cAMP in the absence (▪) or presence (□) of the PKA-I antagonist Rp-8-Br-cAMPS (1 mM, added 0.5 h before DNR). The data are given as mean±S.E.M., n=3. Note that much more N6-Bz-cAMP is required to protect against death in the presence of Rp-8-Br-cAMPS. The significance of the pairwise difference (± Rp-analog at each concentration of N6-Bz-cAMP) was evaluated by Student t-test: **P<0.01, *P<0.05
) or presence (○,▵) of N6-Bz-8-PIP-cAMP (70 μM), which cooperates with 2-Cl-8-HA-cAMP to activate PKA-I, but not PKA-II. The percentage of dead cells dropped more rapidly when 2-Cl-8-HA-cAMP was combined with N6-Bz-8-PIP-cAMP (horizontal arrows), indicating that selective activation of PKA-I could protect both NB4V0 and NB4RIIKD cells against DNR. To better assess the protective synergy between the two analogs (horizontal arrow) the initial cell death was set to 100%. The data represent the average of two experiments, and the original data are shown in Supplementary Figure S2a,b. Similar results were obtained with other PKA-I synergizing analog combinations (data not shown). The enhanced protection with N6-Bz-8-PIP-cAMP was judged by Wilcoxon paired comparison test for the 2-Cl-8-HA-cAMP concentration range 30–600 μM, and was found significant (P<0.01) both for the NB4V0 and the NB4RIIKD cells. (c) NB4V0 cells were treated as described in a, except that the concentration of Sp-5,6-DCl-cBIMPS was varied in the absence (●) or presence (○) of N6-MBC-cAMP (8.5 μM), in order to test for PKA-II synergism. The data represent the average of two experiments (Supplementary Figure S2c). Similar results (no synergy) were obtained with other PKA-II synergizing analog combinations (data not shown). (d) NB4WT cells were treated for 4 h with DNR (5 μM) and increasing concentrations of N6-Bz-cAMP in the absence (▪) or presence (□) of the PKA-I antagonist Rp-8-Br-cAMPS (1 mM, added 0.5 h before DNR). The data are given as mean±S.E.M., n=3. Note that much more N6-Bz-cAMP is required to protect against death in the presence of Rp-8-Br-cAMPS. The significance of the pairwise difference (± Rp-analog at each concentration of N6-Bz-cAMP) was evaluated by Student t-test: **P<0.01, *P<0.05
The phosphorylation state of Bad and CREB in leukemic cells exposed to DNR and cAMP agonists
PKA has a vast number of direct substrates in the intact cell.2, 27 Some of these are convergence points between PKA and kinases involved in general survival signaling, such as ERK and members of the PI3-kinase cascade. They may therefore be particularly important for cell survival. Examples are phosphorylation of Serine 75 and 118 in the BH3-only protein Bad28, 29 (S112 and S155 in murine Bad) and Serine 133 of the transcription factor and AML oncogene CREB.30, 31
We found that cAMP stimulation increased S118Bad phosphorylation in both NB4 cells and APL3 patient blasts (Figures 7a and b; Supplementary Figure S3a). The S118 phosphorylation liberates Bcl-2/Bcl-Xl sequestered by Bad.29 The phosphorylation of S75 of Bad was high already in non-stimulated NB4 cells (Figure 7a). Bad was presumably constitutively phosphorylated also on S99 in NB4 cells, as their Bad migrated more slowly (apparent MW≈20 kDa) upon SDS gel electrophoresis than expected from its MW of 18.5 kDa (Figure 7a, upper lane), a feature typical for P-S75/S99Bad.28, 32

The phosphorylation of Bad and CREB in cAMP- and DNR-exposed leukemic cells. (a) NB4 cells were treated with DNR (5 μM) for 3 h alone or in combination with PGE2/IBMX (2 μM/0.15 mM), and extracts analyzed by immunoblotting for Bad, P-Ser118Bad or P-Ser75Bad, with actin as loading control. The migration corresponding to proteins of Mw 20 and 18.5 kDa is based on interpolation of the migration of relevant standard proteins. (b) Blasts isolated from a patient diagnosed with APL (APL3, Supplementary Table S1) were treated in vitro for 3 h with PGE2/IBMX (2 μM/0.3 mM). Cell extracts were analyzed by immunoblotting for Bad and P-Ser118Bad. (c) HL60 cells were treated for 3 or 6 h with DNR (5 μM) alone or in combination with PGE2/IBMX (2 μM/0.15 mM), and cell extracts were analyzed by immunoblotting for Bad and P-Ser118Bad. (d) Mv4-11 cells were treated for 5 h with DNR (0.4 μM) alone or in combination with PGE2/IBMX (2 μM/0.2 mM), and cell extracts were analyzed by immunoblotting for Bad and P-Ser118Bad. (e) The NB4 cell death was scored by microscopy after incubation with various concentrations of the BH3 mimetic HA14-1, and DNR (3 μM) and PGE2/IBMX (2 μM/0.15 mM). For the left subpanel, the cells were incubated for 3.5 h with 10 μM HA14-1 either alone or during the last 3 h with DNR, PGE2/IBMX or both. For the right hand subpanel, the incubation time with HA14-1 (4 or 6 μM) was 9.5 h, during the last 9 h with DNR, PGE2/IBMX or both. Data are shown as mean±S.E.M., n=3–6. Significance by Student t-test: **P<0.01, *P<0.05. (f) Phospho-protein analysis by flow cytometry for CREB, Erk1/2, Akt, STAT1, STAT5 and STAT3 (see Materials and Methods for detail of phospho-residue detected). NB4 cells were incubated for 1 or 3 h with DNR (5 μM), the PKA agonist N6-Bz-cAMP (0.4 mM), the Epac agonist 8-CPT-2′-Me-cAMP (0.5 mM) or the cAMP-elevating agents PGE2/IBMX (2 μM/0.1 mM). The phospho-epitope labeling intensity is shown as histograms (left subpanels) and heat maps (right subpanels). The control sample was used as a baseline for the heat maps. See Materials and Methods for further experimental details. (g) Phospho-protein analysis by flow cytometry for CREB in blasts isolated from three patients (APL3, AML3 and AML4; Supplementary Table S1). Blasts were treated in vitro for 3 h with PGE2/IBMX (2 μM/0.3 mM) or N6-Bz-cAMP (0.6 mM) before fixation and analysis by flow cytometry. The phospho-epitope labeling intensity is shown as histograms (left subpanels) and heat maps (right subpanels). The control sample was used as a baseline for the heat maps
HL60, Mv4-11 AML cells and AML3 patient blasts were not protected by cAMP (Figures 2a, b and i) and did not show any cAMP stimulation of S118Bad phosphorylation. In the HL60 and Mv4-11 cells, S118Bad was highly phosphorylated already in the resting state (Figures 7c and d), and if anything, P-S118Bad decreased slightly upon prolonged incubation with DNR and PGE2/IBMX (Figures 7c and d). The AML3 blasts had very low Bad expression and P-S118Bad was below the detection limit even with strong cAMP stimulation (data not shown).
The net effect of the cAMP-stimulated S118Bad phosphorylation can be modulated if cAMP also affects the expression of other Bcl-2 family members. cAMP-stimulated NB4, HL60 or Mv4-11 cells had unaltered level of Bax, Bcl-2 and Mcl-1 (Supplementary Figures S3 and S4), whereas DNR caused a rapid decrease of Mcl-1 (Supplementary Figures S3b,c and S4a–c). This suggests that DNR may promote death in all cell lines studied by downregulating Mcl-1.
The consequence of the Mcl-1 downregulation can be compensated by release of Bcl-2 from its complex with Bad in cells showing cAMP-induced S118Bad phosphorylation, like NB4 cells and APL blasts. The BH3-mimetic HA14-1 blocks the ability of Bcl-2 to bind and neutralize Bax33 and should therefore abolish the beneficial effect of S118Bad phosphorylation. At maximally effective concentration (10 μM), HA14-1 induced strong NB4 cell death, which was only moderately antagonized by cAMP (Figure 7e, left subpanel). This is compatible with much of the protective action of cAMP being mediated by S118Bad phosphorylation. At lower HA14-1 concentration (4 or 6 μM) only moderate cell death was observed (Figure 7e, right panel), presumably because only a proportion of the Bcl-2 is occupied by HA14-1. Under such conditions also the Bcl-2 liberated from Bad through S118Bad phosphorylation will be incompletely occupied by HA14-1, which may explain why cAMP provided robust protection against HA14-1 at 4 and 6 μM (Figure 7e, right panel).
cAMP provided some protection against DNR also in the presence of 10 μM HA14-1 (Figure 7e, left panel), suggesting that cAMP may protect also via other mechanisms than Bad phosphorylation. A search for additional survival-associated potential PKA targets by quantitative cell phospho-protein flow cytometry34 revealed increased phosphorylation of the PKA substrate site Ser133 of CREB in NB4 cells incubated with the PKA-directed activators PGE2/IBMX and N6-Bz-cAMP, but not with the Epac-directed activator 8-CPT-2′-Me-cAMP (Figure 7f). We tested next the cAMP-protected APL3 and AML4 and the cAMP-nonresponsive AML3 patient blasts (Figures 2f, i and j) for cAMP-induced P-S133CREB. Only in the two cAMP-protected blasts did cAMP increase P-CREB (Figure 7g). The broadly acting cyclin-dependent protein kinase (CDK) 7/9 inhibitor roscovitine (RCV) blocks transcription stimulated by CREB in a rat AML cell line13 and induces death through transcriptional inhibition in human neutrophils.35 In the NB4 cells, RCV attenuated the protective effect of cAMP stimulators against DNR (Supplementary Figure S5) without affecting the phosphorylation of Bad (data not shown). This supported the existence of protective actions of cAMP involving CDK-dependent events, possibly mediated via PKA activation of CREB.
Several other survival-related protein phosphorylation events were studied, but were not modulated by cAMP. ERK1/2 and PKB/Akt showed no activatory phosphorylation (Figure 7f). The STAT transcription factors showed unaltered phosphorylation both at sites associated with poor (P-STAT5,34 P-STAT336) and favorable (P-STAT134) treatment response in AML (Figure 7f). Furthermore, DNR or cAMP stimulation both failed to affect the phosphorylation of the mTOR substrates p70 S6-kinase and 4E-BP1 (Supplementary Figure S6).
We considered also that clinical, cytogenetic or other parameters available for the APL/AML patients and their blasts might give clues to why only the AML3 blasts failed to be protected by cAMP. The AML3 patient has low-risk AML based on cytogenetics (inv8) and the clinical response (full response to induction therapy including DNR). The low-risk status alone does not explain the lack of cAMP response, as other blasts with low-risk cytogenetics Inv8 (AML1 and AML4) and t8, 37 (AML6) are sensitive to cAMP (Figure 2, Supplementary Table S1). Both NPM1, p53 and Flt3, are wild type in AML3, like for the cAMP-responsive APL3 and AML2 (Supplementary Table S1). Wild-type p53, found important for cAMP to protect lymphoblasts against death,38 was expressed both in the cAMP-resistant AML3 blasts and Mv4-11 cells (see legend to Supplementary Table S1 for details). In conclusion, cAMP protection of DNR-exposed NB4 cells and APL and AML blasts was associated with inactivating phosphorylation of Bad and activating phosphorylation of CREB.
Discussion
As cAMP stimulates ATRA-induced APL blast maturation,3, 4, 5 cAMP agonists have been considered as adjuncts to APL therapy.6, 7, 8 In current APL therapy, an anthracycline is combined with ATRA to eradicate APL blasts resisting ATRA-induced maturation.15, 39 We show here that cAMP agonists accelerate APL progression in mice carrying the NB4 APL cell line and protect APL patient blasts and NB4 cells against anthracycline-induced death in vitro. Also the ATRA-subsensitive (NB4-LR1) and ATRA-insensitive (NB4-LR2) APL cells were protected by cAMP against DNR. The expected beneficial effect of cAMP on APL cell maturation may therefore be outweighed by boosted survival of ATRA-resistant APL blasts.
The cAMP protected not only against the multi-targeting anthracycline drugs,40, 41 but also the glucose metabolism antagonist 2-deoxyglucose, hypoxia/rotenone and ER stress/misfolded protein inducers (the proteasome inhibitor bortezomib and the HSP90 inhibitor geldanamycin). This may explain why cAMP promoted APL progression also in mice not treated with DNR, and suggests that a natural increase of bone marrow cAMP, as may occur as a consequence of inflammation-induced PGE2 production42 can affect APL cell survival also in the absence of specific treatment.
The protective effect of cAMP appeared to be mediated by PKA rather than Epac, based on the efficiency of cAMP analogs stimulating selectively either PKA or Epac. Although the two PKA isozymes (PKA-I and PKA-II) share the same catalytic subunit, they may serve distinct functions due to differential cAMP sensitivity and cellular localization.24, 25 The strictly perinuclear PKA-II may have a localized role not directly related to NB4 cell survival, as PKA-II-directed cAMP analog combinations failed to protect against DNR, and cAMP was fully protective in PKA-II-depleted cells (Figures 5 and 6). In contrast, cAMP analog pairs synergizing for PKA-I activation also synergized for protection against DNR, and the protection afforded by a general PKA-activating cAMP analog was counteracted by the PKA-I-directed compound Rp-8-Br-cAMPS. That PKA-I protects against death is not unprecedented. A PKA isozyme switch from PKA-II to PKA-I is associated with both cellular hypertrophy and the malignant state (see Doskeland et al.43 for review), and activation of PKA-I can protect mature neutrophils against death.10 The possibility that PKA-I modulates mTOR phosphorylation and thereby its activity44 appears unlikely as the mTOR substrates p70 S6-kinase and 4E-BP1 were unaffected by cAMP in the NB4 cells (Supplementary Figure S6).
A search for prosurvival effects of PKA activation in the cAMP-protected NB4 and APL blasts pointed to increased phosphorylation of S118Bad and S133CREB, both of which are recognized PKA substrate sites.29, 31 PKA can thereby turn off the Bcl-2/Bcl-Xl sequestering pro-apoptotic function of Bad45 and activate the AML oncogene and drug resistance factor CREB.31 In HL60 and Mv4-11 cells, where cAMP did not confer protection, the pro-apoptotic action of Bad appeared to be constitutively turned off, as the level of PS118 Bad was high and not modulated further by cAMP elevation. A number of other factors with potential to modulate cell death were studied, but none was regulated by cAMP or correlated with cAMP protection against DNR. Thus, cAMP-stimulated NB4 cells showed unaltered phosphorylation of ERK, STAT family members and mTOR kinase substrates. Furthermore, the non-protected AML3 blasts resembled many cAMP-protected APL/AML blasts regarding clinical parameters, cytogenetics and expression of prognosis-related molecules, like Flt-3, p53 wild type and NPM1 (Supplementary Table S1).
The general survival-promotion expected from Bad phosphorylation and CREB activation27, 29 help explain the broad cAMP-mediated protection of NB4 cells against a panel of cell stressors. Both Bad and CREB phosphorylation are under current investigation as therapeutic targets in AML and other malignancies.46 The antagonism of the cAMP protection by a BH3-only mimetic underscores further the importance and potential druggability of the Bcl-2 family to relieve the cAMP protection of APL cells.
Materials and Methods
Materials
The following substances were from Sigma-Aldrich (Inc., St Louis, MO, USA): DNR, IDA, cycloheximide, RCV, 2-deoxyglucose, geldanamycin, rotenone, ATRA, PGE2, the BH3 mimetic HA14-1, and the PDE inhibitors IBMX, rolipram and milrinone. Bortezomib was from Millenium Pharmaceuticals, Inc (Cambridge, MA, USA). The stabilized PGE2 analog dmPGE2 was from Cayman Chemical (Ann Arbor, MI, USA). The PDE inhibitor theophylline (i.e., its precursor aminophylline) was from Nycomed (Asker, Norway). The cAMP analogs N6-benzoyl-cAMP, (N6-Bz-cAMP), Rp-8-Br-cAMPS, 8-(4-chlorophenylthio)-2′-O-methyl-cAMP (8-CPT-2′-Me-cAMP), Sp-5,6-DCl-cBIMPS, N6-mono-tert-butylcarbamoyl-cAMP (N6-MBC-cAMP), 8-NH-CH3-cAMP, N6-MB-cAMP, 8-CPT-cAMP, 8-PIP-cAMP and N6-benzoyl-8-piperidino-cAMP (N6-Bz-8-PIP-cAMP) were from Biolog Life Science Institute (Bremen, Germany). The synthesis of the new cAMP analog 2-Cl-8-hexylamino-cAMP (2-Cl-8-HA-cAMP) was performed as previously described for the synthesis of the related 2-Cl-8-AHA-cAMP.13
Origin of the cells used
NB4, NB4-LR1 and NB4-LR2 cells were isolated and grown as described.3, 47 NB4 cells were grown in RPMI 1640 (Sigma-Aldrich) with 10% fetal bovine serum (Gibco, Grant Island, NY, USA), L-glutamine (2 mM), streptomycin (5 μg/ml) and penicillin (5 U/ml). Mv4-11 and HL60 cells were from ATTC. Mv4-11 cells were grown in Iscove’s modified Dulbecco’s media (Sigma-Aldrich Inc.) and HL60 in RPMI 1640 supplemented with 10% fetal bovine serum (Gibco), L-glutamine (2 mM), streptomycin (5 μg/ml) and penicillin (5 U/ml). The collection of patient cells was approved by the Regional Research Ethics Committee (Health Region III, Bergen, Norway) and conducted in accordance with the Declaration of Helsinki Principles. For details on patients, see Supplementary Table S1. Samples were collected from patients shortly after the diagnosis of APL or AML, with informed consent, and stored in bio-banks approved by the Norwegian Ministry of Health and the Directorate for Health and Social Affairs. Primary peripheral blood mononuclear cells were isolated by density gradient separation (Ficoll-Hypaque; NyCoMed, Oslo, Norway); the percentage of leukemic cells after gradient separation exceeded 95% for all patients. Cells were stored frozen in liquid nitrogen.48 After thawing, the blasts were suspended in Stem Span medium (Stem Cell Technologies, Vancouver, BC, Canada) before incubation with agents to be tested for death induction.
Viral knockdown of PKA-RIIα and PKA-RIIβ
Knockdown of PKA-RIIα
NB4 cells were transduced with a lentiviral plasmid (pLKO.1/shRNA/PKAR2A, Sigma, CSHGLYC-TRCN0000037806) driving the expression of a short hairpin RNA targeting the sequence:5′- GCTGAGACCTATAACCCTGAT -3′. The empty vector pLKO.1/TRC, given by D. E. Root (MIT, Cambridge, MA, USA)49 was used as control (NB4V0 cells). Lentiviral particle production, NB4 cell infection and selection of transduced NB4 cells were as described.50
Knockdown of PKA-RIIβ
NB4wt and the NB4/pLKO.1/shRNA/PKAR2A subclone with most robust PKA-RIIα protein knockdown were silenced for PKA-RIIβ expression by the PRKA-RIIβ targeting sequence (hairpin loop in small letters) 5′-GGCCTTAATGTACAATACACCAGAGCAGgaccaggaccagCTGCTCTGGGTGTATTGTACATTAAGGCC-3′ inserted in the retroviral vector RRI-Green/L071 (provided by Dr. D. Micklem and Dr. J. Lorens, University of Bergen, Norway) downstream of a modified human U6 promoter. The one mismatch (underlined) in the nonspecific strand served to improve the stability and efficacy of the hairpin. L071/shRNA/PKARIIB and L071/empty constructs were transfected into the Phoenix retroviral packaging cell line to produce viral supernatants harvested 2 days after transfection. Infection of NB4 and NB4/pLKO.1/TRC cells was in the presence of 4 μg/ml of protamine sulfate. Infected cells were sorted 9 days after infection based on their GFP fluorescence.
The NB4 orthotropic xenograft model of APL
Male NOD-scid IL2rγnull (NSG) breeding mice were a generous gift from Dr. L. D. Shultz (Jackson Laboratories, Bar Harbor, Maine, USA16). The animals (7- to 8-week old) received 5 × 106 NB4 cells (Day 0) via the tail vein. On days 4, 5 and 6 post-transplantation, each animal received one intravenous (0.1 ml i.v.), and two intraperitoneal (0.2 ml i.p.) injections, one 5 min before and one 40 min after the i.v. injection. Group 1 (Ctr, 5 animals) received phosphate-buffered saline (PBS) both i.v. and i.p. Group 2 (‘cAMP’, 6 animals) received PBS i.v. and a mixture of dmPGE2 (to 125 μg/kg) and aminophylline (to 20 mg/kg) i.p. Group 3 (‘DNR’, 6 animals) received DNR (diluted in physiological saline to give 2.5 mg/kg) i.v. and PBS i.p. Group 4 (‘DNR+cAMP’, 6 animals) received both DNR and the dmPGE2/aminophylline mixture.
The health status and weight of the mice was monitored and moribund animals were humanely euthanized. The animal experiments were approved by The Norwegian Animal Research Authority and performed in accordance with The European Convention for the Protection of Vertebrates Used for Scientific Purposes. The survival data are presented according to Kaplan and Meier and survival distributions analyzed by the Mantel-Cox log rank statistics, using PASW Statistics (version 18.0).
Assessment of cell death and determination of cAMP content
Cells (at 0.5 × 106/ml) were exposed to various cell death-modulating agents. Apoptosis of cell lines was assessed mainly by microscopy to visualize surface budding and nuclear chromatin condensation. Upon prolonged incubation with apoptogens, some cells underwent secondary necrosis. Although swollen, such cells retained enough of the apoptotic phenotype to be recognized as ‘dead’. In most cases, the % of apoptotic+(secondarily) necrotic cells is given as % dead cells. NB4 cell lines were also studied by flow cytometry on a FASC Accuri C6 (BD Biosciences, San Jose, CA, USA, Accuri Cytometry), after staining with Alexa Fluor 647-Annexin V (Molecular Probes, Grand Island, NY, USA) and propidium iodide (PI), and the forward and side scatter determined. At least 50 000 non-gated live cell events were collected for each sample. The determination of fluorescence from fluorescence-labeled Annexin V (binds to phosphatidyl serine) is a challenge for cells treated with DNR, which has a strong fluorescence when excited by the 488-nm blue argon laser. We eliminated the problem by using Annexin V coupled to the far-red Alexa Fluor 647 dye, which was excited with a 635-nm red diode and sampled in the FL-4 channel. PI gives a stronger signal than DNR when excited at 488 nm, particularly when sampled after a long-pass 670-nm filter (FL-3). The DNR content was determined after excitation at 488 nm and sampling in the FL-1 channel.
For primary human blasts at least 30 000 non-gated events were collected for each sample. For the first two samples studied (APL1,2) staining was with PI, and % PI-positive cells taken as % death. For all other samples cell death was determined based on the sum of Annexin V+PI-positive cells. It should be noted that the primary blasts were more prone to (secondary) necrotic death than the cell lines and only minor differences were observed using PI-positive and Annexin V+PI-positive cells.
For study under hypoxic conditions, the medium was pre-equilibrated for 24 h in 2% O2, 5% CO2 atmosphere. For statistical analysis, Wilcoxon Paired Comparison test and Student’s t-Test were used.
The ATRA-induced differentiation of NB4 cells was evaluated by the nitroblue tetrazolium reduction assay as described.47
Determination of cAMP:
The cell medium was rapidly removed and cAMP extracted in 5% trichloroacetic acid with 0.1 M HCl. The trichloroacetic acid was extracted with acetone and the extract neutralized in phosphate-HEPES buffer before determination of cAMP by a competitive binding assay based on displacement of [3H]cAMP from the RIα subunit of PKA.37
Immunoblotting and immunocytofluorescence
Immunoblotting was as described.17 Proteins were detected with antibodies against, P-Ser155Bad (murine, corresponds to human P-Ser118), Mcl-1, BAX, Pro-caspase 3, PKA-RIIα (Santa Cruz, Biotechnology, Dallas, TX, USA), Bad, Bcl-2, PKA-RIα, PKA-RIIβ (BD Biosciences), P-Ser112Bad (murine, corresponds to human P-Ser75; Cell Signaling, Danvers, MA, USA), p23 (mouse monoclonal JJ3, generously supplied by Dr. D. O. Toft, Mayo Clinic, Rochester, MN, USA) or β-actin (Amersham Biosciences, Piscataway, NJ, USA). For immunofluorescence, cells were collected by centrifugation, fixed in 2% PFA in PBS (10 min), permeabilized in 0.1% Triton X-100 in PBS (10 min) and blocked (2% BSA in PBS, 1 h). Incubation with anti-PKA-RI and anti-PKA-RII (in PBS-BSA) was overnight. After washing in PBS, cells were incubated 1 h with secondary antibody Alexa 488-IgG or Alexa 594-IgG (Invitrogen), washed and mounted on slides with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA, USA). The cell fluorescence was analyzed on a Leica DMRD microscope.
Intracellular phospho-epitope staining for flow cytometry
Phospho-flow cytometry analysis was performed as previously described.34 Alexa 647-conjugated anti-phospho-CREB (pS133), anti-phospho-STAT1 (pY701), anti-phospho-STAT5 (pY694), anti-phospho-STAT3 (pS727), anti-phospho-Erk1/2 (pT202/pY204) and anti-phospho-Akt (pS473) were from BD Biosciences. At least 30 000 non-gated live cell events were collected for each sample on a FACS Calibur (Becton Dickinson, Franklin Lakes, NJ). Normalization of flow cytometry data was performed by comparing median fluorescence intensity values to an unstimulated control using the log10 ratio of a sample divided by the control. With this scale an increment of +1 corresponds to a 10-fold increase in median fluorescence intensity.
Abbreviations
- APL:
-
acute promyelocytic leukemia
- ATRA:
-
all-trans retinoic acid
- cAMP:
-
cyclic AMP
- dmPGE2:
-
16,16-dimethyl PGE2
- DNR:
-
daunorubicin
- IBMX:
-
3-isobutyl-1-methylxanthine
- IDA:
-
idarubicin
- PDE:
-
phosphodiesterase
- PGE2:
-
prostaglandin E2
- RCV:
-
roscovitine
References
Bos JL . Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci 2006; 31: 680–686.
Kopperud R, Krakstad C, Selheim F, Doskeland SO . cAMP effector mechanisms. Novel twists for an ‘old’ signaling system. FEBS Lett 2003; 546: 121–126.
Ruchaud S, Duprez E, Gendron MC, Houge G, Genieser HG, Jastorff B et al. Two distinctly regulated events, priming and triggering, during retinoid-induced maturation and resistance of NB4 promyelocytic leukemia cell line. Proc Natl Acad Sci USA 1994; 91: 8428–8432.
Habib A, Hamade E, Mahfouz R, Nasrallah MS, de The H, Bazarbachi A . Arsenic trioxide inhibits ATRA-induced prostaglandin E2 and cyclooxygenase-1 in NB4 cells, a model of acute promyelocytic leukemia. Leukemia 2008; 22: 1125–1130.
Roussel MJ, Lanotte M . Maturation sensitive and resistant t(15;17) NB4 cell lines as tools for APL physiopathology: nomenclature of cells and repertory of their known genetic alterations and phenotypes. Oncogene 2001; 20: 7287–7291.
Parrella E, Gianni M, Cecconi V, Nigro E, Barzago MM, Rambaldi A et al. Phosphodiesterase IV inhibition by piclamilast potentiates the cytodifferentiating action of retinoids in myeloid leukemia cells. Cross-talk between the cAMP and the retinoic acid signaling pathways. J Biol Chem 2004; 279: 42026–42040.
Guillemin MC, Raffoux E, Vitoux D, Kogan S, Soilihi H, Lallemand-Breitenbach V et al. In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J Exp Med 2002; 196: 1373–1380.
Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat Med 2008; 14: 1333–1342.
Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG . Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun 2010; 2: 216–227.
Krakstad C, Christensen AE, Doskeland SO . cAMP protects neutrophils against TNF-{alpha}-induced apoptosis by activation of cAMP-dependent protein kinase, independently of exchange protein directly activated by cAMP (Epac). J Leukoc Biol 2004; 76: 641–647.
Geering B, Simon HU . Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ 2011; 18: 1457–1469.
Garcia-Bermejo L, Perez C, Vilaboa NE, de Blas E, Aller P . cAMP increasing agents attenuate the generation of apoptosis by etoposide in promonocytic leukemia cells. J Cell Sci 1998; 111 (Pt 5): 637–644.
Huseby S, Gausdal G, Keen TJ, Kjaerland E, Krakstad C, Myhren L et al. Cyclic AMP induces IPC leukemia cell apoptosis via CRE-and CDK-dependent Bim transcription. Cell Death Dis 2011; 2: e237.
Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009; 113: 1875–1891.
Sanz MA, Montesinos P, Rayon C, Holowiecka A, de la Serna J, Milone G et al. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood 2010; 115: 5137–5146.
Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005; 174: 6477–6489.
Gausdal G, Gjertsen BT, Fladmark KE, Demol H, Vandekerckhove J, Doskeland SO . Caspase-dependent, geldanamycin-enhanced cleavage of co-chaperone p23 in leukemic apoptosis. Leukemia 2004; 18: 1989–1996.
Pesic M, Podolski A, Rakic L, Ruzdijic S . Purine analogs sensitize the multidrug resistant cell line (NCI-H460/R) to doxorubicin and stimulate the cell growth inhibitory effect of verapamil. Invest New Drugs 2010; 28: 482–492.
Chandra P, Zhang P, Brouwer KL . Short-term regulation of multidrug resistance-associated protein 3 in rat and human hepatocytes. Am J Physiol Gastrointest Liver Physiol 2005; 288: G1252–G1258.
Gewirtz DA . A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57: 727–741.
Benito J, Shi Y, Szymanska B, Carol H, Boehm I, Lu H et al. Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS One 6: e23108.
Burnett A, Wetzler M, Lowenberg B . Therapeutic advances in acute myeloid leukemia. J Clin Oncol 2011; 29: 487–494.
Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK et al. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 2003; 278: 35394–35402.
Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E . Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol 2010; 283: 235–330.
DiPilato LM, Cheng X, Zhang J . Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA 2004; 101: 16513–16518.
Gjertsen BT, Mellgren G, Otten A, Maronde E, Genieser HG, Jastorff B et al. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem 1995; 270: 20599–20607.
Naviglio S, Caraglia M, Abbruzzese A, Chiosi E, Di Gesto D, Marra M et al. Protein kinase A as a biological target in cancer therapy. Expert Opin Ther Targets 2009; 13: 83–92.
Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB et al. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 2000; 6: 41–51.
Danial NN . BAD: undertaker by night, candyman by day. Oncogene 2008; 27 (Suppl 1): S53–S70.
Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell 2005; 7: 351–362.
Gonzalez GA, Montminy MR . Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989; 59: 675–680.
Polzien L, Baljuls A, Rennefahrt UE, Fischer A, Schmitz W, Zahedi RP et al. Identification of novel in vivo phosphorylation sites of the human proapoptotic protein BAD: pore-forming activity of BAD is regulated by phosphorylation. J Biol Chem 2009; 284: 28004–28020.
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA 2000; 97: 7124–7129.
Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 2004; 118: 217–228.
Leitch AE, Lucas CD, Marwick JA, Duffin R, Haslett C, Rossi AG . Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ 2012; 19: 1950–1961.
Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood 2009; 113: 4052–4062.
Ekanger R, Sand TE, Ogreid D, Christoffersen T, Doskeland SO . The separate estimation of cAMP intracellularly bound to the regulatory subunits of protein kinase I and II in glucagon-stimulated rat hepatocytes. J Biol Chem 1985; 260: 3393–3401.
Naderi EH, Findley HW, Ruud E, Blomhoff HK, Naderi S . Activation of cAMP signaling inhibits DNA damage-induced apoptosis in BCP-ALL cells through abrogation of p53 accumulation. Blood 2009; 114: 608–618.
Kamimura T, Miyamoto T, Harada M, Akashi K . Advances in therapies for acute promyelocytic leukemia. Cancer Sci 2011; 102: 1929–1937.
Laurent G, Jaffrezou JP . Signaling pathways activated by daunorubicin. Blood 2001; 98: 913–924.
Gausdal G, Gjertsen BT, McCormack E, Van Damme P, Hovland R, Krakstad C et al. Abolition of stress-induced protein synthesis sensitizes leukemia cells to anthracycline-induced death. Blood 2008; 111: 2866–2877.
Sakata D, Yao C, Narumiya S . Prostaglandin E2, an immunoactivator. J Pharmacol Sci 2010; 112: 1–5.
Doskeland SO, Maronde E, Gjertsen BT . The genetic subtypes of cAMP-dependent protein kinase—functionally different or redundant? Biochim Biophys Acta 1993; 1178: 249–258.
Mavrakis M, Lippincott-Schwartz J, Stratakis CA, Bossis I . mTOR kinase and the regulatory subunit of protein kinase A (PRKAR1A) spatially and functionally interact during autophagosome maturation. Autophagy 2007; 3: 151–153.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Cho EC, Mitton B, Sakamoto KM . CREB and leukemogenesis. Crit Rev Oncog 2011; 16: 37–46.
Duprez E, Ruchaud S, Houge G, Martin-Thouvenin V, Valensi F, Kastner P et al. A retinoid acid ‘resistant’ t(15;17) acute promyelocytic leukemia cell line: isolation, morphological, immunological, and molecular features. Leukemia 1992; 6: 1281–1287.
Bruserud O, Gjertsen BT, Foss B, Huang TS . New strategies in the treatment of acute myelogenous leukemia (AML): in vitro culture of aml cells--the present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001; 19: 1–11.
Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006; 124: 1283–1298.
Pendino F, Nguyen E, Jonassen I, Dysvik B, Azouz A, Lanotte M et al. Functional involvement of RINF, retinoid-inducible nuclear factor (CXXC5), in normal and tumoral human myelopoiesis. Blood 2009; 113: 3172–3181.
Acknowledgements
This study was supported by the Norwegian Cancer Society, Norske Kvinners Sanitetsforening, The Association pour la Recherche contre le Cancer, The Fondation de France, Cent pour Sang la Vie, Marie-Curie Actions and by Aurora program. The expert technical assistance of Nina Lied Larsen, Khanh K. Dao, Sjur Huseby, Siri Strømsøy, Lene Vikebø and Mihaela Popa is highly appreciated.
Author contributions
GG, AW, and SOD. designed the research, performed most experiments, analyzed data and wrote the paper. EN, FP, ML and ES-B provided the NB4-LR1 and -LR2 sub-clones and generated the NB4-RIIKD cell line, characterized their content and distribution of PKA subunits, and contributed to data interpretation and writing. UH and FS contributed to syntheses of cAMP analogs. JS, BTG and ØB performed phospho-flow analysis, provided and characterized patient blasts, and helped presenting data. NR and EM designed and performed animal experiments. RK and LH contributed to data analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Gausdal, G., Wergeland, A., Skavland, J. et al. Cyclic AMP can promote APL progression and protect myeloid leukemia cells against anthracycline-induced apoptosis. Cell Death Dis 4, e516 (2013). https://doi.org/10.1038/cddis.2013.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.39
Keywords
This article is cited by
-
cAMP-PKA/EPAC signaling and cancer: the interplay in tumor microenvironment
Journal of Hematology & Oncology (2024)
-
A forward genetic screen identifies modifiers of rocaglate responsiveness
Scientific Reports (2021)
-
Meta-analysis of gene signatures and key pathways indicates suppression of JNK pathway as a regulator of chemo-resistance in AML
Scientific Reports (2021)
-
Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer
Nature Medicine (2015)
-
Dual effect of oxidative stress on leukemia cancer induction and treatment
Journal of Experimental & Clinical Cancer Research (2014)
