
Abstract
BTBD10, an Akt interactor, activates Akt by decreasing the protein phosphatase 2A-mediated dephosphorylation and inactivation of Akt. Overexpression of BTBD10 suppresses motor neuron death that is induced by a familial amyotrophic lateral sclerosis (ALS)-linked superoxide dismutase 1 (SOD1) mutant, G93A-SOD1 in vitro. In this study, we further investigated the BTBD10-mediated suppression of motor neuron death. We found that the small interfering RNA-mediated inhibition of BTBD10 expression led to the death of cultured motor neurons. In Caenorhabditis elegans (C. elegans), disruption of the btbd-10 gene caused not only loss of neurons, including both motor and touch-receptor neurons, but also a locomotion defect. In addition, we found that the expression of BTBD10 was generally decreased in the motor neurons from patients of sporadic ALS and transgenic mice overexpressing G93A-SOD1 (G93A-SOD1-transgenic mice). Collectively, these results suggest that the reduced expression of BTBD10 leads to motor neuron death both in vitro and in vivo.
Similar content being viewed by others


Main
A gain-of-toxic-function of an intracellular molecule or a loss of normal function of an essential intracellular molecule, including an intracellular prosurvival signal, contributes to cell death. The Akt family of proteins (Akt1, 2 and 3 in mammalian cells) provides a major intracellular prosurvival signal by phosphorylating many target proteins.1 The major upstream activator of Akt is the phosphatidylinositol 3-kinase (PI3K)-mediated signal that activates Akt by phosphorylation. Akt's activity is also regulated by protein phosphatase-mediated dephosphorylation.2 In our previous study, we showed that BTBD10 activates Akt by inhibiting protein phosphatase 2A (PP2A)-mediated Akt dephosphorylation.3
Recent advances in genetics and neuroscience have suggested that both gains of toxic and/or losses of normal function of intracellular molecules contribute to the pathogenesis of neurodegenerative diseases. Amyotrophic lateral sclerosis (ALS) is a representative motor neuron-specific neurodegenerative disease.4 Approximately 10% of ALS cases are genetically inherited.4, 5 Dominant mutations in the superoxide dismutase 1 (SOD1) gene (the first identified familial ALS (FALS)-linked gene) account for approximately 20% of FALS cases. The gain-of-toxic-function of SOD1, caused by a FALS-linked mutation, is thought to be closely linked to the ALS pathogenesis. The second identified FALS-linked gene, a FALS-linked mutation of which results in an autosomal-recessive phenotype, encodes a protein named alsin.6, 7 Loss of alsin function, caused by a FALS-linked mutation, leads to the abnormal endolysosomal trafficking and contributes to motor neuron death.8 We have independently shown that wild-type alsin suppresses mutant SOD1-induced neuronal cell death by activating a prosurvival pathway involving Rac1, PI3K and Akt3, and that FALS-linked mutations in the gene abolish the alsin function.9, 10
To further characterize the Akt-mediated prosurvival pathways that are linked to the ALS pathogenesis, we previously performed an yeast-two hybrid analysis and identified BTBD10 as an Akt3-binding protein. BTBD10 activates Akt by inhibiting PP2A-mediated Akt dephosphorylation and inactivation.3 BTBD10 also contributes to the insulin-stimulated phosphorylation of Akt and pancreatic beta cell proliferation.11 We further found that overexpression of BTBD10 inhibited FALS-linked mutant SOD1-induced motor neuron death in vitro.3 Although these findings together suggest that BTBD10 is an upstream regulator of Akt that may be involved in the survival of motor neurons, an in vivo relationship between BTBD10 malfunction and motor neuron death has yet to be established.
To address this issue, we decided to disrupt the btbd-10 gene in Caenorhabditis elegans (C. elegans) and observe the effect of the disruption of the btbd-10 gene on survival of motor neurons. We chose this approach for two reasons. First, the BTBD10 gene is highly conserved in C. elegans (Supplementary Figure S1) and C. elegans has only one ortholog of the human BTBD10 gene. Second, recent studies have shown that transgenic overexpression of FALS-linked G85R-SOD1 causes presynaptic dysfunction and a locomotion defect in C. elegans12 and that transgenic overexpression of TAR DNA-binding protein-43 (TDP-43) or fused in sarcoma (FUS) induces both motor neuron death and a locomotion defect in C. elegans.13, 14 As TDP-43 and FUS, both of which were RNA/DNA-binding proteins, have been recently identified as major components of cytoplasmic ubiquitinated inclusion bodies in most ALS cases, including FALS and sporadic ALS (SALS) cases, and various missense mutations of TDP-43 and FUS cause FALS in an autosomal-dominant fashion,5, 15, 16 it is hypothesized that the gain-of-toxic-function and/or the loss of normal function of TDP-43 or FUS is linked to motor neuron death. These results together suggest that C. elegans can be used as an animal model of human motor neuron diseases.
In this study, we show that the reduction in the BTBD10 level led to the death of cultured motor neurons, that disruption of the btbd-10 gene caused loss of neurons and impairment of motor function in C. elegans, and that the BTBD10 levels were reduced in the motor neurons in spinal cords from SALS patients and in G93A-SOD1-transgenic mice. Taken together, these results suggest that reduced expression of BTBD10 is closely linked to motor neuron death.
Results
Reduction of BTBD10 expression causes motor neuron death in vitro
In our previous study, we showed that the overexpression of BTBD10 inhibited the G93A-SOD1-induced death in cultured neuronal cells.3 Reciprocally, in this study, we found that small interfering RNA (siRNA)-mediated reduction of BTBD10 expression increased cleavage of caspase-3 and poly (ADP-ribose) polymerase (PARP) in NSC34 motor neuron cells (Figure 1a). Reduced expression of BTBD10 also increased the proportion of NSC34 motor neuron cells that belonged to the subG1 population, which represents the fraction of dead cells (Figure 1b).

siRNA-mediated reduction of BTBD10 expression causes cell death and caspase activation. (a and b) NSC34 motor neuron cells, transfected with each of two independent siRNAs for BTBD10, were harvested for immunoblot analysis with indicated antibodies (a) and for FACS analysis to determine the increase in cell death, estimated by the proportion of cells belonging to the sub-G1 population (b). Statistical significance was determined by ANOVA followed by Fisher’s PLSD test (*P<0.001, the error bars indicate S.D.)
Disruption of the btbd-10 gene causes loss of touch-receptor neurons in C. elegans
The amino-acid sequence of BTBD10 is highly conserved among Homo sapiens, Mus musculus and C. elegans (Supplementary Figure S1). The C-terminal 330 amino-acid region of BTBD10 is the most highly conserved region (Supplementary Figure S1) and is essential for the interaction with Akt family proteins.3 To examine the role of BTBD10 in neuronal cell survival in vivo, we isolated two C. elegans lines, named tm3335 or tm3607, bearing artificially mutated btbd-10 genes. tm3335 contains a 297-base pair (bp) deletion plus a 5-bp insertion while tm3607 contains a 207-bp deletion in the middle of exon 4 (Supplementary Figure S2). Both deletions were predicted to cause a frameshift and a premature termination of the btbd-10 gene, which should give rise to mutants of btbd-10 in which the C-terminal region (approximately 250 amino acids) was deleted.
We first examined the effect of the disruption of the btbd-10 gene on touch-receptor neurons by crossing these lines with a C. elegans line, bzIs8, in which six touch-receptor neurons were labeled with green fluorescent protein (GFP) (Figure 2a).17 We confirmed that BTBD10 was ubiquitously expressed including the touch-receptor neurons using transgenic C. elegans bearing Pbtbd−10::mCherry::btbd-10, which expresses an mCherry-tagged worm BTBD10 (red) under the worm btbd-10 gene promoter (Figure 2b). In young-adult (4 days old) btbd-10(tm3335);bzIs8 and btbd-10(tm3607);bzIs8 worms, the 6 touch-receptor neurons were randomly lost and approximately 10% of the worms lost at least 1 touch-receptor neuron (Figure 2c). The frequency of the loss of at least 1 touch-receptor neuron (abbreviated ‘loss of touch-receptor neurons’) in these btbd-10 mutants was comparable to the frequency of the loss of touch-receptor neurons in tat-1(tm1034);bzIs8 worms, a previously reported line that exhibits substantial loss of touch-receptor neurons (Figure 2c).18 Importantly, the loss of touch-receptor neurons in btbd-10 mutants was rescued by the introduction of the 8.07-kb btbd-10(+) transgene (Figure 2c and Supplementary Figure S2) or by the introduction of the cDNA encoding N-terminally mCherry-tagged human BTBD10 under the control of the worm btbd-10 gene promoter (Pbtbd−10::mCherry::hBTBD10) (Figure 2c). Loss of touch-receptor neurons was also suppressed by the disruption of the ced-3 gene (Figure 2c), an executioner of the caspase cascade in C. elegans.19

BTBD10 has an essential role in the survival of touch-receptor neurons in young-adult C. elegans. (a) Touch-receptor neurons in C. elegans. PLMR and PLML touch-receptor neurons appeared merged in this particular picture. (b) Pictures of one of the touch-receptor neurons in btbd-10(tm3335);bzIs8 worms carrying extrachoromosomal array containing Pbtbd10::mCherry::worm btbd-10. The mCherry-tagged worm BTBD10 (red) was expressed ubiquitously in C. elegans including the touch-receptor neurons expressing GFP protein under the control of the mec-4 promoter (green). (c) The ratios of young-adult worms (4 days old) lacking at least 1 touch-receptor neurons were calculated for each C. elegans line
These results support the hypothesis that the disruption of the btbd-10 gene promotes the death of touch-receptor neurons in young-adult (4 days old) worm via the activation of the caspase cascade in C. elegans. However, the worm's touch sensation was not apparently affected by the loss of a single touch-receptor neuron and there were only a few cases in which more than one neuron was lost.
BTBD10 functions upstream of Akt in C. elegans
BTBD10 increased Akt phosphorylation by inhibiting PP2A-mediated Akt dephosphorylation.3 Akt family proteins (Akt1, Akt2 and Akt3 in mammalian cells and akt-1 and akt-2 in C. elegans) inhibit apoptosis by activating a variety of target molecules.1 Although germ cells derived from akt-1 or akt-2 loss-of-function mutants are more sensitive to DNA damage than wild-type worms,20 the effect of reduced Akt expression on the neuronal cell survival has not been examined. We examined whether neuronal cell death occurred in akt-1 or akt-2 mutant worms. Approximately 6.5–8.5% of akt-1 mutants (ok525;bzIs8 or tm399;bzIs8) or akt-2 mutants (ok393 bzIs8 or tm1075 bzIs8) lost touch-receptor neurons (Figure 2c). Next, we crossed an akt-1 or akt-2 mutant with a btbd-10 mutant and observed that the disruption of the btbd-10 gene did not affect the frequency of the loss of touch-receptor neurons in akt-1 or akt-2 mutants (Figure 2c). Moreover, the introduction of the constitutively active form of human Akt1 under the control of the akt-1 gene promoter (Pakt−1::cahAkt-1::mCherry) reduced the percentage of the loss of touch-receptor neurons from 10.2 to 2.2% in tm3335;bzIs8, from 6.9 to 3.2% in akt-1;bzIs8, and from 8.0 to 2.3% in akt-2 bzIs8 (Figure 2c). These results collectively indicate that Akt behaves as a downstream effector of BTBD10 signaling in C. elegans.
Disruption of the btbd-10 gene causes loss of motor neurons and a locomotion defect in young-adult C. elegans
We next examined the effects of the disruption of the btbd-10 gene on the number of two types of motor neurons that innervate dorsal muscle named dorsal B-motor neurons (DB) and dorsal A-motor neurons (DA). These cholinergic motor neurons are responsible for forward and backward locomotion, respectively. We crossed tm3335 and tm3607 with a C. elegans line, named evls82b, in which DB and DA neurons were labeled with the unc-129::GFP reporter (Figure 3a).21 In young-adult (4 days old) btbd-10(tm3335);evls82 and btbd-10(tm3607);evls82 worms, 14 DA and DB motor neurons were randomly lost and 6.7% and 7.8% of worms lost at least one DB and DA motor neurons, respectively, whereas no evls82 worms lost DB and DA neurons (Figure 3b). We also noted that 4.8% of btbd-10(tm3335);evls82 and 2.9% of btbd-10(tm3607);evls82 young-adult worms had at least one mislocalized DB or DA motor neurons, whereas no evls82 worms contained a mislocalized neuron (Figure 3c). The mislocalization of neurons was observed only in the two neurons that are located near the vulva. This result indicates that the disruption of the btbd-10 gene promotes death and mislocalization of DB and DA motor neurons in young-adult worms.

The disruption of the btbd-10 gene causes a loss or a mislocalization of motor neurons and a locomotion defect in young-adult C. elegans. (a) DB and DA motor neurons expressing GFP in wild-type (evls82) and mutant [btbd-10(tm3335);evls82] C. elegans. One neuron (arrow) disappeared in the mutant worm. (b) The ratios of young-adult worms (4 days old) lacking at least one DA and DB motor neurons were calculated for each C. elegans line. (c) In the left panel, mislocalized neurons were shown. Closed arrows indicate mislocalized neurons and open arrow indicates the point that the mislocalized neuron should have been localized. In the right panel, the ratios of young-adult worms (4 days old) lacking at least mislocalized DA and DB motor neurons were calculated for each C. elegans line. (d) One minute after setting a worm in 1 ml of M9 medium, body bending was counted for 1 min. Statistical significance was determined by ANOVA followed by Bonferroni’s Multiple Comparison Test (*P<0.0001, the error bars indicate S.E.M.)
To examine the locomotion defect that was putatively caused by the loss of motor neurons, we compared the frequency of body bending (thrash) between btbd-10(tm3335 or tm3607) and N2 young-adult worms. We observed a significant decrease in the frequency of body bending in btbd-10 mutant worms, compared with N2 worms (Figure 3d and Supplementary movies). Importantly, this locomotion defect was completely rescued by the introduction of the btbd-10(+) transgene (Figure 3d). These results together suggest that expression of BTBD10 is essential for the survival of motor neurons and for motor performance in C. elegans.
We also examined the effect of the disruption of the btbd-10 gene on the GABAergic motor neurons named dorsal D (DD) and ventral D (VD). In C. elegans adults, sinusoidal motion depends on a signaling network in which cholinergic motor neurons alternately excite dorsal and ventral muscles as GABAergic innervation concurrently inhibits contraction on the opposite side. We crossed tm3335 with a C. elegans line with 6 DD and 13 VD neurons that were labeled by the unc-25::GFP reporter, named juIs76 (Figure 4a).22 None of these 19 GABAergic motor neurons were lost from young-adult (4 days old) btbd-10(tm3335) juIs76 worms (Figure 4a). However, 17 days after birth, 25% of btbd-10(tm3335) juIs76 worms lost at least 1 D-type motor neurons, whereas 9.8% of juIs76 worms lost D-type neurons (Figure 4b). Six hermaphrodite-specific VC neurons, which extend synapses onto egg-laying muscles and neurons, were labeled by the pes-10::GFP reporter, named vsIs13 (Figure 4c).23 In all, 4% and 6% of young-adult vsIs13 and tm3335;vsIs13 worms lost at least one VC neuron, respectively (Figure 4c). However, at the age of 17 days, 51.7% of vsIs13 worms lost at least 1 VC neurons and 84% of tm3335;vsIs13 worms lost at least 1 VC neuron (Figure 4d). These results together suggest that the disruption of the btbd-10 gene also promotes motor neuronal death during senescence. It is also hypothesized that a larger portion of VC neurons than that of DD and VD neurons was lost from aged worms possibly because their presence is not essential after the worms’ reproductive period ends.

The disruption of the btbd-10 gene promotes a loss of DD and VD motor neurons in aged C. elegans. (a and b) The ratios of young adult worms (4 days old) (a) or aged worms (17 days old) (b) lacking at least 1 DD and VD GABAergic motor neurons were calculated for each C. elegans line. juIs76 indicates worms with DD and VD neurons expressing GFP. (c and d) The ratios of young adult (c) or aged worms (d) lacking at least 1 VC motor neurons were calculated for each C. elegans line. vsIs13 indicates worms with VC neurons expressing GFP. tm3335 indicates worms deficient in the btbd-10 gene
BTBD10 expression is reduced in motor neurons in ALS
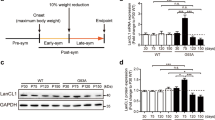
BTBD10 was highly expressed in motor neurons in the anterior horn of the spinal cord and only minimally expressed in astrocytes (Supplementary Figures S3A and B). ALS is the representative motor neuron-specific degenerative disease. We therefore chose to examine whether BTBD10 expression was altered in motor neurons in the spinal cords of ALS patients. We compared the number of BTBD10-positive motor neurons between SALS patients and non-ALS controls. BTBD10-positive motor neurons were defined as motor neurons with apparent BTBD10 positivity, the staining intensities of which were clearly stronger than the staining in the background neuropil surrounding motor neurons. Their detailed clinical characteristics and the numbers of analyzed motor neurons are shown in Table 1. Microscopic analysis indicated that the numbers of BTBD10-positive motor neurons were significantly decreased in the surviving motor neurons in the lamina 9 of autopsied SALS cases (Figures 5a and b and Table 1). We also observed that the reduction in BTBD10 expression occurred more frequently in large motor neurons mainly consisting of α motor neurons or lower motor neurons than in small neurons (data not shown) and that the reduction in the number of BTBD10-positive neurons in the ALS patients became more marked as their disease duration increased (Table 1). These results suggested that the reduced expression of BTBD10 is a feature of SALS.

Decreased BTBD10 expression in motor neurons of ALS patients. (a) Paraffin sections of spinal cords from SALS patients or non-ALS controls were stained with a BTBD10 antibody (brown-dark brown). Areas, surrounded by dashed lines, represent spinal ventral horns. Arrowheads indicate the remaining motor neurons. (b) The numbers of BTBD10-positive motor neurons in the inner area, outer area, or all area of lamina 9 of anterior horns of the human spinal cords were counted and the median percentage of BTBD10-positive motor neurons in all samples (control (n = 6) (filled circle), SALS (n = 6) (closed circle) ) was calculated. The average of the ratio of BTBD10-positive neuron was indicated as a crossbar. The percentage of BTBD10-positive motor neurons was significantly decreased in SALS (P<0.05 by χ2 test)
We also examined motor neurons of G93A-SOD1-transgenic mice,24 which began to exhibit impairment of motor function at approximately 100 days old and died at approximately 140 days old.25 Compared with wild-type littermates, they began to show a gradual reduction in the number of motor neurons in the anterior horn at 120 days (Supplementary Figure S3C). Similarly, BTBD10 expression began to gradually decline at 120 days in the surviving neurons of G93A-SOD1 mice (Supplementary Figure S3C).
Discussion
In our previous study, we showed that overexpression of BTBD10 inhibited G93A-SOD1-induced death of cultured motor neurons.3 In this study, we further showed that the loss of BTBD10 led to the death of NSC34 motor neurons (Figure 1). More importantly, the disruption of the btbd-10 gene caused the loss of motor neurons in C. elegans during multiple phases of their lives (Figures 3 and 4). Together, these data indicate that BTBD10 has an essential role in preventing motor neuron death both in vitro and in vivo. Therefore, the insufficiency in BTBD10 function is thought to lead to motor neuron death.
BTBD10 activates Akt by inhibiting PP2A-mediated Akt dephosphorylation and inactivation in vitro.3 In this study, we recapitulated these data in vivo by showing that BTBD10 functions upstream of Akt in C. elegans (Figure 2). As the expression of constitutively active human Akt-1 almost completely rescued the neuronal loss, caused by the deletion of the btbd-10 gene, it is highly likely that the Akt family of proteins is a major downstream target of BTBD10 in C. elegans. However, apart from the Akt family proteins, BTBD10 may have multiple downstream targets, with which it may interact through BTBD10's broad complex, tramtrack (ttk) and bric-a-brac/poxvirus and zinc-finger proteins-homologous domain.3 In fact, we have detected multiple BTBD10-binding proteins other than Akt and PP2A (unpublished observation).
Multiple studies have shown that impaired Akt-mediated prosurvival signal contributes to motor neuron death. The level of phosphorylated Akt, the activated form of Akt, is downregulated in motor neurons in autopsied spinal cords of FALS patients and SALS patients,26 and the activation of the PI3K/Akt prosurvival pathway delays the progression of ALS-linked motor neuron death in vivo.27, 28, 29 In this study, consistent with these previous observations, we found that the BTBD10 levels were decreased in the motor neurons of the spinal cords from SALS patients (Figure 5b). Although there was one ALS patient (case 5, see Table 1), whose motor neurons displayed no reduction in the BTBD10 level, the overall findings suggest that reduced expression of BTBD10 may contribute to motor neuron death in most SALS cases by downregulating the Akt-mediated prosurvival signal. However, it remains unclear whether these reductions in the BTBD10 levels were sufficient for motor neuron death in ALS. More importantly, there is no direct evidence supporting this hypothesis thus far. Therefore, it is possible that the observed reduction in the BTBD10 level might be not a cause but a result or an accidental coincidence of motor neuron death.
How was the BTBD10 levels decreased in motor neurons of ALS patients? The gain-of-toxic and/or loss of normal function of FUS or TDP-43 may contribute to the pathogenesis of SALS and FALS.5, 15, 16 In this study, we further found that the adenoviral vector-mediated low-grade overexpression of human FUS reduced both BTBD10 expression and the Akt phosphorylation at Ser473 (which correlates with Akt activation) (Supplementary Figure S4), whereas the adenovirus-mediated low-grade overexpression of TDP-43 showed a minimal tendency to reduce BTBD10 expression in some human cultured cells (Supplementary Figure S5). The overexpression of FUS by only ∼1.5–2-fold more than the endogenous level is enough to reduce the BTBD10 level. As the overexpression of each protein is thought to mimic its gain-of-toxic-function state,30, 31 these results suggest that the gain-of-function of FUS may contribute to the downregulation of BTBD10 in the SALS patients. However, additional studies to confirm this observation are needed before a conclusion can be drawn.
In this study, we found that BTBD10 expression was reduced in motor neurons in mice constitutively overexpressing G93A-SOD1 (Supplementary Figure S3C). This result suggests that BTBD10 is also a downstream target of the gain-of-toxic-function state of SOD1. However, an independent group has reported that, inconsistent with our results, the Akt phosphorylation was not attenuated in the motor neurons in the spinal cords of G93A-SOD1-transgenic mice.32 One possible explanation that reconciles these findings with the findings in this study is that BTBD10 has downstream effectors besides Akt that may mediate BTBD10's prosurvival activity in G93A-SOD1-transgenic mice. In fact, we identified some BH3-only proteins as non-Akt BTBD10 target candidates (unpublished observation). If this hypothesis is correct, the mechanism underlying the BTBD10-induced protection against cell death is more complicated than originally thought.
We also found that expression of the btbd-10 gene prevented the loss of touch-receptor neurons (Figure 2). This result suggests that sensory neurons may be lost when the BTBD10 level is reduced. We examined the BTBD10 level in the sensory neurons of the spinal cords from the ALS patients. In contrast to the motor neurons, the BTBD10 levels in the sensory neurons were not different between the ALS patients and the normal controls (data not shown). This result is consistent with the general notion that the function of sensory neurons is unperturbed in ALS patients. Finally, we observed that the deletion of the btbd-10 gene did not apparently shorten the life span or change the appearance of C. elegans (data not shown) although we did not systematically explore the effect of the BTBD10 loss on non-neuronal cells. It is therefore unlikely that the loss of the btbd-10 gene causes a major defect in non-neuronal cells.
In summary, we found that reduced expression of BTBD10 was toxic to motor neurons (and to touch-receptor neurons) and that the BTBD10 level was reduced in motor neurons from ALS patients. Although the mechanism underlying decreased BTBD10 expression in motor neurons of ALS patients and the link between decreased BTBD10 expression and motor neuron death in ALS still must be clarified, these findings provide suggestive evidence for the mechanism underlying motor neuron death.
Materials and Methods
Cell lines
NSC34 cells, HeLa cells, and HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Wako Pure Chemical Industries, Osaka, Japan) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), penicillin/streptomycin solution (Invitrogen, Carlsbad, CA, USA). NTERA-2 (NT-2) cells were cultured in DMEM/Ham's F12 mixture (Wako Pure Chemical Industries), supplemented with 10% fetal bovine serum (Hyclone), penicillin/streptomycin (Invitrogen).
Gene silencing
Gene silencing of BTBD-10 was performed according to a plasmid-based siRNA method,33 as described previously.3
Adenoviral vector-mediated expression
The adenovirus expression system was purchased from TaKaRa (Shiga, Japan). The FUS cDNA was inserted into the Swa I site of a cosmid adenoviral vector, pAxCALNLw. In this vector, a stuffer DNA fragment, sandwiched by two loxP sequences, is located just upstream of the cloned cDNA and interferes with its expression. If an adenovirus expressing cre-recombinase is co-introduced into the cells, the stuffer is removed and FUS is expressed. The TDP-43 cDNA was inserted into the Swa I site of a cosmid adenoviral vector, pAxCAwt, for the expression of genes without co-expression of cre-recombinase. All viruses were grown in HEK293 cells and purified by double CsCl2 gradient ultracentrifugation and infection was performed by adding recombinant adenoviruses to serum-containing media, as previously described.34 The infected cells were harvested at 48 h for HeLa cells or 32 h for NT2 cells after infection just before the cells started to become dead.
Quantitative real-time PCR
HeLa cells were co-infected with adenoviruses encoding cre-wt-FUS or LacZ together with cre-recombinase or LacZ and harvested at 48 h after infection for total RNA purification using ISOGEN (Nippon gene, Tokyo, Japan). First-strand cDNA was synthesized from 1 μg of total RNA using the PrimeScript High Fidelity RT-PCR kit (TaKaRa) and DNA fragments of BTBD10, FUS, or glyceraldehyde-3-phosphate dehydrogenase (G3PDH) were amplified with the 7500 Real-Time PCR system using the SYBR green real time PCR kit (Applied Biosystems, Carlsbad, CA, USA) by using the following parameters: 95 °C for 10 min, followed by 40 cycles of 95 °C for 5 s, 55 °C for 10 s, and 72 °C for 34 s. A region of human BTBD10 cDNA was amplified using a sense primer 5′-ACACGACCCAATGAGAAAGG-3′ and an antisense primer 5′-TCATGCATTAGGGCACTGAG-3′. A region of the human FUS cDNA was amplified using a sense primer 5′-CCACGGACACTTCAGGCTAT-3′ and an antisense primer 5′-TACCGTAACTTCCCGAGGTG-3′. A region of human G3PDH cDNA was amplified using a sense primer 5′-GAAGGTGAAGGTCGGAGTC-3′ and an antisense primer 5′-GAAGATGGTGATGGGATTTC-3′. Amplification data were analyzed by instrument software (Applied Biosystems).
Strains
C. elegans strains were maintained using the standard techniques.35 N2 Bristol was used as the wild-type strain. The strain bzIs8[mec-4::gfp] was a kind gift of Dr. Ding Xue. The akt-1 (ok525) and akt-2 (ok393) mutants, OH4125 (evIs82b), CZ1200 (juIs76) and LX959 (vsls13) were obtained from the Caenorhabditis Genetics Center (CGC (University of Minnesota, Mineapolis, MN, USA)). The akt-1 (tm399), akt-2 (tm1075), ced-3 (tm1196) and btbd-10 (tm3335 and tm3607) mutants were isolated as described in detail below. The genetic crossings were confirmed by PCR.
Isolation of the mutated alleles
The mutated alleles were obtained from a trimethylpsoralen/ultraviolet mutagenized library, as described previously36 and identified by PCR amplification and all mutants were backcrossed with N2 at least twice before analysis. Detailed information of primer sets used is available in Supplementary Materials and Methods.
Transgenic worms
Germline transformation was performed as described previously.37 The btbd-10 genome constructs, Pbtbd-10::mCherry::worm btbd-10, or Pbtbd-10::mCherry::hBTBD10 (at 50–100 ng/μl each), were injected into btbd-10(tm3335 or tm3607) worms with the appropriate genetic background along with pRF-4[rol-6d] or Pmyo−2::DsRed (100–150 ng/μl) as an injection marker. For the expression analysis, Pbtbd-10::mCherry::worm btbd-10 (200 ng/μl) was injected into btbd-10 (tm3335) worms. Pakt−1::cahAkt-1::mCherry (200 ng/μl) was injected into worms with the appropriate genetic background.
Analysis of the number of touch-receptor and motor neurons
Young-adult worms (4 days old) were mounted on a 3% agar pad on a cover slip and paralyzed. The number of touch-receptor neurons or motor neurons was determined with a fluorescence microscope (Olympus Optical Co., Ltd, Tokyo, Japan). The percentage of worms lacking at least 1 touch-receptor neuron or motor neurons was calculated.
Body bending assay
Young-adult worms (4 days old) were transferred to an NGM plate (ø=3.5 cm) filled with 1 ml of the M9 buffer. After 1 min of adaptation, the number of sinusoidal curves made during locomotion for 1 min was scored as the number of body bending.38
Antibodies
Polyclonal antibodies to BTBD10 were generated, as previously described.3 Phospho-Akt (Ser473) (#9271), Akt (#9272), cleaved caspase-3 (#9661), caspase-3 (#9662) and cleaved PARP antibodies (#9544) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-actin antibody was purchased from SIGMA (St. Louis, MO, USA). An FUS antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). A TDP-43-N (10782-2-AP) antibody was purchased from ProteinTech Group, Inc. (Chicago, IL, USA).
Transgenic mice
All experimental procedures were approved by the Institutional Animal Experiment Committee of Tokyo Medical University. Transgenic mice expressing a high copy number of human Gly-93-Ala SOD1 (G93A-SOD1) were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and maintained in a specific pathogen-free environment, as previously described.39
Cell cycle analysis
NSC34 cells, into which siRNA for mouse BTBD10 was transfected, were harvested at 72 h after transfection. Cell cycle determination was performed by FACS analysis with FACS Calibur (Becton-Dickinson, Franklin Lakes, NJ, USA). The percentage of cells belonging to sub-G1 DNA content was determined with the MODFIT program.
Human spinal cord samples
Experiments using human samples were approved by the Institutional Ethic Committee of Tokyo Medical University. Informed consent for the experimental study using each human spinal cord sample was obtained after the nature and possible consequences of the studies were explained. The lumbar or thoracic spinal cord samples from 6 SALS patients (average age: 68.5-year old, 4 males, 2 females) were diagnosed as ALS by clinical and neuropathological criteria or six non-ALS controls (average age: 71.8-year old, 3 men, 3 females) with no pathologic findings in the spinal cord (Table 1). Spinal cords were fixed with 4% paraformaldehyde in phosphate-buffered solution (pH7.4) and embedded in paraffin. Five-micrometers thick transverse paraffin sections were prepared for immunohistochemistry. The intervals from death to the start of autopsies were within 3 h, as shown in the Table 1.
Immunohistochemistry
Frozen sections of spinal cord from G93A-SOD1-transgenic mice or their littermates were immunostained with the primary anti-BTBD10 antibody (1 : 50) and the secondly FITC-conjugated goat anti-rabbit IgG antibody (1 : 200) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Sections were also probed with mouse monoclonal antibody to GFAP (Cell Signaling Technology) or mouse monoclonal antibody to non-phosphorylated neurofilaments (SMI32) (Sternberger Monoclonals, Baltimore, MD, USA) and visualized with a Texas-Red conjugate goat anti-mouse IgG antibody. Nuclei were stained with Hoechst 33258 (SIGMA). The sections were imaged using the Zeiss LSM510 confocal microscope (Carl Zeiss, Tokyo, Japan). Human spinal cord sections were deparaffinized and incubated with an antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA) at 70 °C for 15 min for antigen retrieval. The sections were washed with Tris-buffered saline containing 0.1% Tween 20 and incubated for 30 min with 0.3% H2O2 in methanol to quench endogenous peroxidase activity followed by blocking the sections with normal goat serum. Sections were incubated with the primary anti-BTBD10 antibody (1 : 50) in Tris-buffered saline with 0.1% Tween-20 (TBST) at 4 °C for 5 days. Detection was performed with the Vectastain Elite ABC Kit (rabbit IgG) (Vector Laboratories) and the Vector DAB peroxidase substrate kit (Vector Laboratories). BTBD10-positive motor neurons were defined as motor neurons with apparent BTBD10 positivity, the staining intensities of which were clearly stronger than the staining in the background neuropil surrounding motor neurons. There are two types of motor neurons in the spinal cords; large neurons consisting of α motor neurons that are responsible for skeletal muscle contraction and small neurons. We counted both neurons. A cell was counted as a neuron if the length of the cell body was longer than 15 μm, the length of the cytoplasm was larger than the nucleus, and the cell did not show a star-like shape.
Abbreviations
- ALS:
-
amyotrophic lateral sclerosis
- C. elegans :
-
Caenorhabditis elegans
- FALS:
-
familial ALS
- FUS:
-
fused in sarcoma
- G3PDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- GFP:
-
green fluorescent protein
- PI3K:
-
phosphatidylinositol 3-kinase
- PARP:
-
poly (ADP-ribose) polymerase
- PP2A:
-
protein phosphatase 2A
- SALS:
-
sporadic ALS
- siRNA:
-
small interfering RNA
- SOD1:
-
superoxide dismutase 1
- TDP-43:
-
TAR DNA-binding protein-43
References
Franke TF . PI3K/Akt: getting it right matters. Oncogene 2008; 27: 6473–6488.
Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K et al. Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol 2004; 24: 8778–8789.
Nawa M, Kanekura K, Hashimoto Y, Aiso S, Matsuoka M . A novel Akt/PKB-interacting protein promotes cell adhesion and inhibits familial amyotrophic lateral sclerosis-linked mutant SOD1-induced neuronal death via inhibition of PP2A-mediated dephosphorylation of Akt/PKB. Cell Signal 2008; 20: 493–505.
Rothstein JD . Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 2009; 65 (Suppl 1): S3–S9.
Pasinelli P, Brown RH . Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006; 7: 710–723.
Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 2001; 29: 166–173.
Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 2001; 29: 160–165.
Hadano S, Otomo A, Kunita R, Suzuki-Utsunomiya K, Akatsuka A, Koike M et al. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One 2010; 5: e9805.
Kanekura K, Hashimoto Y, Niikura T, Aiso S, Matsuoka M, Nishimoto I . Alsin, the product of ALS2 gene, suppresses SOD1 mutant neurotoxicity through RhoGEF domain by interacting with SOD1 mutants. J Biol Chem 2004; 279: 19247–19256.
Kanekura K, Hashimoto Y, Kita Y, Sasabe J, Aiso S, Nishimoto I et al. A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J Biol Chem 2005; 280: 4532–4543.
Wang X, Liu Y, Yang Z, Zhang Z, Zhou W, Ye Z et al. Glucose metabolism-related protein 1 (GMRP1) regulates pancreatic beta cell proliferation and apoptosis via activation of Akt signalling pathway in rats and mice. Diabetologia 2011; 54: 852–863.
Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet 2009; 5: e1000350.
Liachko NF, Guthrie CR, Kraemer BC . Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci 2010; 30: 16208–16219.
Murakami T, Yang SP, Xie L, Kawano T, Fu D, Mukai A et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum Mol Genet 2012; 21: 1–9.
Lagier-Tourenne C, Polymenidou M, Cleveland DW . TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 2010; 19: R46–R64.
Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 2010; 67: 739–748.
Royal DC, Bianchi L, Royal MA, Lizzio Jr M, Mukherjee G, Nunez YO et al. Temperature-sensitive mutant of the Caenorhabditis elegans neurotoxic MEC-4(d) DEG/ENaC channel identifies a site required for trafficking or surface maintenance. J Biol Chem 2005; 280: 41976–41986.
Darland-Ransom M, Wang X, Sun CL, Mapes J, Gengyo-Ando K, Mitani S et al. Role of C. elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science 2008; 320: 528–531.
Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO . Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 1999; 126: 1011–1022.
Quevedo C, Kaplan DR, Derry WB . AKT-1 regulates DNA-damage-induced germline apoptosis in C. elegans. Curr Biol 2007; 17: 286–292.
Nash B, Calarita A, Zheng H, Roy PJ, Culotti JG . The Forkhead transcription factor UNC-130 is required for the graded spatial expression of the UNC-129 TGF-β guidance factor in C. elegans. Genes Dev 2000; 14: 2486–2500.
Huang X, Cheng HJ, Tessier-Lavigne, Jim Y . MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron 2002; 34: 563–576.
Bany IA, Dong MQ, Koelle MR . Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J Neurosci 2003; 23: 8060–8069.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994; 264: 1772–1775.
Chiba T, Hashimoto Y, Tajima H, Yamada M, Kato R, Niikura T et al. Neuroprotective effect of activity-dependent neurotrophic factor against toxicity from familial amyotrophic lateral sclerosis-linked mutant SOD1 in vitro and in vivo. J Neurosci Res 2004; 78: 542–552.
Dewil M, Lambrechts D, Sciot R, Shaw PJ, Ince PG, Robberecht W et al. Vascular endothelial growth factor counteracts the loss of phospho-Akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 2007; 33: 499–509.
Tolosa L, Mir M, Olmos G, Lladó J . Vascular endothelial growth factor protects motoneurons from serum deprivation-induced cell death through phosphatidylinositol 3-kinase-mediated p38 mitogen-activated protein kinase inhibition. Neuroscience 2009; 158: 1348–1355.
Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004; 429: 413–417.
Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG et al. Control of motoneuron survival by angiogenin. J Neurosci 2008; 28: 14056–14061.
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 2010; 107: 3858–3863.
Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet 2011; 7: e1002011.
Peviani M, Cheroni C, Troglio F, Quarto M, Pelicci G, Bendotti C . Lack of changes in the PI3K/AKT survival pathway in the spinal cord motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 2007; 34: 592–602.
Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC et al. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 5515–5520.
Tsuji K, Mizumoto K, Yamochi T, Nishimoto I, Matsuoka M . Differential effect of ik3-1/cables on p53- and p73-induced cell death. J Biol Chem 2002; 277: 2951–2957.
Brenner S . The genetics of Canorhabditis elegans. Genetics 1974; 77: 71–94.
Gengyo-Ando K, Mitani S . Characterization of mutations induced by ethyl methanesulfonate, UV, and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem Biophys Res Commun 2000; 269: 64–69.
Mello CC, Kramer JM, Stinchcomb D, Ambros V . Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 1991; 10: 3959–3970.
Lee H, Cho JS, Lambacher N, Lee J, Lee SJ, Lee TH et al. The Caenorhabditis elegans AMP-activated protein kinase AAK-2 is phosphorylated by LKB1 and is required for resistance to oxidative stress and for normal motility and foraging behavior. J Biol Chem 2008; 283: 14988–14993.
Chiba T, Yamada M, Hashimoto Y, Sato M, Sasabe J, Kita Y et al. Development of a femtomolar-acting humanin derivative named colivelin by attaching activity-dependent neurotrophic factor to its N terminus: characterization of colivelin-mediated neuroprotection against Alzheimer's disease-relevant insults in vitro and in vivo. J Neurosci 2005; 25: 10252–10261.
Acknowledgements
We thank Dr. D Xue for providing worm strains used in this study, Dr. RY Tsien for providing mCherry cDNA and T Hiraki for essential assistance throughout this study. This work was supported in part by the Nakabayashi Trust for ALS research (to M.N.).
Author Contributions
MN designed and performed most experiments. EK-N designed and co-performed worm experiments. SM designed and supervised worm experiments. SA contributed to this study as an expert of ALS. KO performed immunohistochemical analysis. MM conceived, designed, and supervised the study. MM, MN, and EK-N wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by L Greene
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Nawa, M., Kage-Nakadai, E., Aiso, S. et al. Reduced expression of BTBD10, an Akt activator, leads to motor neuron death. Cell Death Differ 19, 1398–1407 (2012). https://doi.org/10.1038/cdd.2012.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.19
Keywords
This article is cited by
-
An ancient role for collier/Olf/Ebf (COE)-type transcription factors in axial motor neuron development
Neural Development (2019)
-
Specific Induction of Akt3 in Spinal Cord Motor Neurons is Neuroprotective in a Mouse Model of Familial Amyotrophic Lateral Sclerosis
Molecular Neurobiology (2014)
-
KCTD20, a relative of BTBD10, is a positive regulator of Akt
BMC Biochemistry (2013)
