
Abstract
Transmembrane BAX inhibitor motif-containing (TMBIM)-6, also known as BAX-inhibitor 1 (BI-1), is an anti-apoptotic protein that belongs to a putative family of highly conserved and poorly characterized genes. Here we report the function of TMBIM3/GRINA in the control of cell death by endoplasmic reticulum (ER) stress. Tmbim3 mRNA levels are strongly upregulated in cellular and animal models of ER stress, controlled by the PERK signaling branch of the unfolded protein response. TMBIM3/GRINA synergies with TMBIM6/BI-1 in the modulation of ER calcium homeostasis and apoptosis, associated with physical interactions with inositol trisphosphate receptors. Loss-of-function studies in D. melanogaster demonstrated that TMBIM3/GRINA and TMBIM6/BI-1 have synergistic activities against ER stress in vivo. Similarly, manipulation of TMBIM3/GRINA levels in zebrafish embryos revealed an essential role in the control of apoptosis during neuronal development and in experimental models of ER stress. These findings suggest the existence of a conserved group of functionally related cell death regulators across species beyond the BCL-2 family of proteins operating at the ER membrane.
Similar content being viewed by others


Main
Apoptosis is a conserved cell death mechanism essential for normal development and tissue homeostasis in multicellular organisms. In mammals, the BCL-2 family of proteins is a group of crucial upstream regulators of the caspase cascade, comprising both pro- and anti-apoptotic components.1 Although apoptosis is observed in most multicellular organisms, the BCL-2 family of proteins as a whole is poorly conserved in invertebrates including worms, flies, and other species.1, 2 In fact, only two BCL-2 homologues are present in flies with controversial roles in programmed cell death.3, 4
A pioneering screening to identify human genes that prevents BAX toxicity in a yeast assay identified transmembrane BAX inhibitor motif-containing (TMBIM)-6, also known as BAX inhibitor-1 (BI-1), as a new mammalian gene that negatively regulates apoptosis (reviewed in Robinson KS et al. and Reimers K et al.5, 6). Further studies demonstrated that TMBIM6/BI-1 is a six transmembrane-spanning protein, located at the endoplasmic reticulum (ER) that has a relevant role in preventing apoptosis.5 Remarkably, bioinformatic analysis defined a putative family of at least six highly conserved orthologs of TMBIM6/BI-1containing the consensus motif UPF0005 with unknown function, a domain encoding for six to seven transmembrane-spanning regions.7 TMBIM family of proteins includes the founder member TMBIM6/BI-1, TMBIM1/RECS1 (responsive to centrifugal force and shear stress gene 1 protein), TMBIM2/LFG (life guard), TMBIM3/GRINA (glutamate receptor ionotropic NMDA protein 1), TMBIM4/GAAP (Golgi anti-apoptotic-associated protein), and TMBIM5/GHTIM (growth hormone-inducible transmembrane protein). The TMBIM family of proteins is highly conserved in mammals, zebrafish, and flies, with homologs present even in species where no BCL-2 family members have been identified, including plants, yeast and many viruses.7
TMBIM6/BI-1 is the best characterized component of the TMBIM family of proteins, having anti-apoptotic activity in cellular and mouse models of ER stress.8, 9 At the molecular level, TMBIM6/BI-1 regulates ER calcium homeostasis, impacting stimulated calcium release and calcium-mediated cell death (see examples in Chae et al.8 and Xu Q et al.10). TMBIM6 has been also recently shown to modulate autophagy levels under conditions of ER stress and nutrient starvation.11 ER stress is a process that involves the accumulation of misfolded proteins in the ER lumen.12 Successful adaptation to ER stress depends on the engagement of the unfolded protein response (UPR), an integrated signal transduction pathway that reduces unfolded protein load.13 The UPR is initiated by the activation of three major stress sensors termed PERK, IRE1α, and ATF6, which in concert govern the expression of a large spectrum of partially overlapping target genes to recover ER homeostasis (reviewed in Hetz C et al.12 and Woehlbier U et al.14).
The majority of TMBIM family members remain poorly characterized, lacking in most cases in vivo verification of their biological functions. Here, we investigated the possible impact of TMBIM3/GRINA in the regulation of apoptosis. Our results uncovered a critical role of TMBIM3/GRINA in the control of cell death in cellular and animal models of ER stress possibly owing to the regulation of ER calcium homeostasis. Our results identify a new conserved regulator of apoptosis that operates in concert with TMBIM6/BI-1 on a core cell death pathway at the ER membrane.
Results
TMBIM3/GRINA expression has a protective activity against ER stress-induced apoptosis
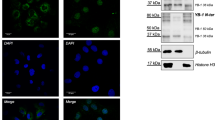
TMBIM3 primary sequence is highly conserved and related to the TMBIM protein family (Supplementary Figures S1a, S1b and S1c). We cloned five TMBIM family members as MYC-tagged proteins, and assessed their subcellular distribution in murine embryonic fibroblasts (MEFs), observing distinct patterns of expression where TMBIM3-MYC colocalized with an ER and Golgi marker (Figure 1a and Supplementary Figure S1d). We then stably expressed TMBIM3-MYC in MEFs and assessed its possible impact on apoptosis (Figure 1b). Cells were stimulated with different concentrations of the ER stress-inducing agents tunicamycin (Tm); blocks N-glycosylation) or thapsigargin (Thg); inhibits the ER-calcium pump SERCA. In addition, we tested the susceptibility of TMBIM3-overexpressing cells to other intrinsic death stimuli non-related to ER stress, including etoposide, staurosporine, nutrient starvation, and serum withdrawal, in addition to tumor necrosis factor-α. Unexpectedly, TMBIM3 expression led to a significant protection against ER stress but not to other death stimuli as monitored by propidium iodide staining and fluorescence-activated cell sorting analysis (Figures 1b and c). These results were confirmed by quantifying the sub-G1 hypodiploid apoptotic population (Figure 1d and Supplementary Figure S2a). These observations were recapitulated in an acute setting of ER stress, where cells were transiently exposed to Tm for 4 h and then replated in normal cell culture media (Figure 1e). No changes in proliferation were observed after overexpressing TMBIM3-MYC (Supplementary Figure S2b). Taken together, these results suggest a specific role of TMBIM3 in the control of ER stress-mediated cell death.

TMBIM3 protects against ER stress-mediated apoptosis. (a) WT MEFs cells were transiently transfected with TMBIM3-MYC or TMBIM6-MYC expression vectors and their subcellular localization was evaluated by immunofluorescence (green) after co-staining with Brefeldine A-bodipy (red) using a confocal microscopy. Nucleus was stained with Hoechst (blue). (b) Left panel: MEFs cells were stably transduced with retroviral expression vectors for TMBIM3-MYC or empty vector (MOCK) and its expression assessed by western blot. Hsp90 levels were monitored as loading control. Right panel: cells were exposed to indicated concentrations of Tm or Thg for 24 h, and cell death was evaluated by propidium iodide (PI) staining and fluorescence-activated cell sorting (FACS) analysis. Mean and S.D. are presented of four independent experiments. Student's t-test was used to analyze statistical significance between control and TMBIM3-overexpressing cells (*P<0.05, **P<0.01, ***P<0.001). (c) Alternatively, cells were analyzed as described in (b) after treatment with 10 ng/ml TNF-α and 10 μM Actinomycin D (TNF-α), 40 μM etoposide (Eto), 0.1 μM stausorporine (Sts), nutrient starved, or exposed to serum withdrawal. Data represents mean and S.D. of four independent experiments. Student's t-test indicated no significant differences between MOCK or TMBIM3-expressing cells for each treatment. (d) Apoptosis was monitored in cells described in (b) after treatment with 1 μg/ml Tm or 1 μM Thg by quantifying the percentage of hypodoploid cells by FACS analysis. Mean and S.D. are presented of three independent experiments. Student's t-test was used to analyze statistical significance between control and TMBIM3-MYC-overexpressing cells (*P<0.05). (e) MOCK and TMBIM3-MYC-expressing cells were exposed to a pulse of 2.5 μg/ml Tm for 4 h. Then Tm was washed out, and cells re-plated into 10 cm cell culture dish and cultured for up to 5 days. Total cell number was visualized by staining with crystal violet (left panel). Number of cells per area (1.5 mm2) was quantified over time. Mean and S.D. are presented of three independent experiments. Student's t-test was used to analyze statistical significance between control and TMBIM3-overexpressing cells (*P<0.05). The color reproduction of this figure is available at Cell Death and Differentiation Journal online
TMBIM3/GRINA and TMBIM6/BI-1 have synergistic anti-apoptotic activity
We then targeted tmbim6 mRNA with two independent small hairpin RNA (shRNA) constructs through lentiviral-mediated delivery (Supplementary Figure S3a). Targeting TMBIM3 in TMBIM6 wild-type (WT) cells did not result in significant spontaneous cell death after transient expression of the shRNA constructs (Figures 2a and b). In sharp contrast, knocking down TMBIM3 in TMBIM6 knockout (KO) cells led to the appearance of classical morphological changes resembling apoptosis, including cell shrinkage and detachment (Figure 2a, and Supplementary Figure S3b), followed by cell death (Figures 2b and c).

Downregulation of TMBIM3 induces apoptosis of TMBIM6-deficient cells. (a) TMBIM6 WT and KO MEFs cells were transiently transduced with lentiviral vectors expressing two different shRNA constructs directed to tmbim3 mRNA (sh#1 and sh#2) or to the luciferase mRNA (shLuc) as control. After 4 days of transduction, cell morphology was visualized by phase contrast microscopy. Results are representative of three independent experiments. (b) Alternatively, cell viability of experiments presented in (a) was quantified by PI staining and FACS analysis. Mean and S.D. are presented of four independent experiments. Student's t-test was used to analyze statistical significance between WT and TMBIM6 KO cells (*P<0.05). (c) In parallel to (a), nuclear morphology was visualized after Hoechst staining. Right panel: quantification of the percentage of cells with condensed and fragmented nucleus. Mean and S.D. are presented of three independent experiments. Student's t-test was used to analyze statistical significance between WT and TMBIM6 KO cells under different conditions (**P<0.01, ***P<0.001). (d) TMBIM6 WT (Left panel) and KO (right panel) cells were stably transduced with lentiviruses expressing shLuc or shRNA constructs against tmbim3 (shTMBIM3) construct #2, and then cells were exposed to indicated concentrations of Tm. After 24 h, cell viability was evaluated by PI staining and FACS analysis. Mean and S.D. are presented of three or four determinations. Statistically significant differences between TMBIM6KO shLuc and shTMBIM3 were detected by multiple comparisons using two-way ANOVA. (e) TMBIM6 WT and KO cells were stably transduced with retroviruses expressing TMBIM3-MYC or MOCK, and then cells were exposed to 1 μg/ml Tm or 1 μM Thg for 24 h. Cell viability/number was measured with the MTS assay. Average and S.D. of three determinations is presented representative of two independent experiments. (f) 293T cells were co-transfected with expression vectors for TMBIM6-human influenza hemagglutinin (HA) and TMBIM3-MYC or empty vector (pCDNA.3). After 48 h, TMBIM3-MYC was immunoprecipitated (IP) and then the co-precipitation of TMBIM6-HA was assessed by western blot (WB) analysis
Next, we addressed the impact of TMBIM3 deficiency on ER stress-mediated cell death by stably transducing cells with shRNA #2. Knocking down TMBIM3 increased the susceptibility of TMBIM6 KO cells to ER stress-inducing agents (Figure 2d, right panel). Interestingly, no significant effects were observed when similar experiments were performed in TMBIM6 WT cells (Figure 2d, left panel). TMBIM3/TMBIM6 double deficiency did not affect the induction of cell death triggered by etoposide, staurosporine, TNF-α, or nutrient starvation (Supplementary Figure S3c and not shown). Moreover, TMBIM3-MYC partially reverted the susceptibility of TMBIM6 deficient cells to Tm or Thg treatment (Figure 2e).
Based on the synergistic effects observed for TMBIM3 and TMBIM6 in the regulation of cell death, we performed immunoprecipitation experiments to assess a possible physical interaction between both proteins. We transiently expressed MYC-tagged TMBIM3 and human influenza hemagglutinin-tagged TMBIM6 in 293T cells and performed immunoprecipitation of TMBIM3-MYC from total protein extracts. We were able to detect a co-immunoprecipitation of TMBIM6 with TMBIM3, indicating that they do form part of a protein complex (Figure 2f).
ER stress triggers the upregulation of TMBIM3/GRINA through a PERK-dependent mechanism
Our results suggest that TMBIM3 expression specifically confers protection against ER stress. Real-time PCR analysis revealed a strong and sustained upregulation of tmbim3 mRNA levels in cells exposed to Tm (>nine fold induction), showing similar kinetics of induction as erdj4 and chop, a classical UPR-target gene (Figure 3a and Supplementary Figure S4b). In contrast, tmbim6 mRNA only showed a slight but not significant increase in its mRNA levels (Figure 3a).

Tmbim3 mRNA levels are regulated by the PERK signaling branch of the UPR. (a) MEFs cells were exposed to 1 μg/ml Tm for indicated time points, and tmbim3, tmbim6 and erdj4 (positive control) mRNA levels were evaluated by real-time PCR and normalized with actin levels. Mean and S.D. are presented of three independent experiments. (b) Right panel: PERK WT and KO cells were exposed to 1 μg/ml Tm for indicated time points, and tmbim3 mRNA levels were evaluated by real-time PCR. (c) In parallel, in the same cells described in (b), xbp-1 mRNA splicing was monitored by RT-PCR (upper panel). PCR fragments corresponding to spliced and unspliced forms are indicated. Bottom panels: CHOP or Hsp90 levels were monitored by western blot in the same experiment. (d) IRE1αKO MEFs cells were stably transduced with retroviruses expressing IRE1α-HA or MOCK, and tmbim3 mRNA levels were monitored by real-time PCR after treatment with Tm for indicated time points. (e) ATF6α WT and KO cells were treated with Tm for 24 h and tmbim3 mRNA levels were monitored as described in (a). As positive control, chop mRNA was monitored. Mean and S.E. is presented of three independent experiments. (f) PERK WT and KO cells were exposed to indicated concentrations of Tm for 24 h, and cell viability was evaluated by PI staining and FACS analysis. Mean and S.D. are presented of four independent experiments. Student's t-test was used to analyze statistical significant differences (**P<0.01; ***P<0.001). (g) MEFs cells stably transduced with MOCK or TMBIM3-MYC expression vector were incubated with 1 μg/ml Tm for 16 h. Then, the expression levels of the ER chaperones and foldases BIP, PDI, ERp74, ERp57, and EDEM was evaluated by western blot. Hsp90 levels were monitored as loading control. Note: The electrophoretic shift in EDEM1 is because of deglycosylation of the protein upon Tm treatment. (h) WT mice were i.p. injected with indicated concentrations of Tm. After 16 h of injection, kidneys were collected. Then, tmbim3, chop, actin, and xbp-1 mRNA splicing levels were monitored by RT-PCR using cDNA prepared from total mRNA. Each well of the gel represents independent animals. (i) atf4+/+ or atf4−/− mice were injected with 100 ng/g Tm. After 16 h, tmbim3 and actin mRNA levels were monitored by semiquantitative PCR in kidney tissue described in (h). Each well represents an independent animal. (j) In parallel, tmbim3 mRNA levels were measured by real-time PCR in the same experiment performed in (i). Mean and S.D. are presented of the analysis of three or four independent animals. Statistically significant differences detected by multiple comparisons using one-way ANOVA and Newman-Keuls Multiple Comparison Test (***P<0.01)
Subsequently, we investigated the possible control of tmbim3 expression by specific UPR-signaling branches. The PERK pathway of the UPR regulates apoptosis-related genes by the induction of the transcription factor ATF4.12 IRE1α is an endoribonuclease that upon activation catalyzes the unconventional splicing of the XBP-1, leading the expression of a potent UPR transcription factor.12 Finally, ATF6 encodes a bZIP transcription that under ER stress conditions translocates to the Golgi, where it is processed releasing the transcription factor.15 Treatment of PERK KO cells with Tm led to almost a complete attenuation in tmbim3 upregulation under ER stress conditions (Figure 3b). As positive and negative control, we monitored CHOP levels and XBP-1 mRNA splicing in PERK KO cells (Figure 3c and Supplementary Figure S4b). We did not observe any significant alteration on tmbim3 mRNA induction in IRE1α-deficient (Figure 3d and Supplementary Figures S4c, d) or ATF6α-deficient (Figure 3e) or knockdown cells (Supplementary Figures S4e and f), suggesting that the PERK signaling branch specifically regulates TMBIM3 expression.
In agreement with early reports indicating a crucial role of PERK in cell survival under ER stress conditions,12 we observed an enhanced susceptibility of PERK KO cells to Tm treatment when compared with WT control cells (Figure 3f). As additional control experiments, we monitored the levels of classical ER stress responsive genes, including ATF4, CHOP, in addition to proteins involved in folding and quality control/endoplasmic reticulum-associated degradation mechanisms, and did not detect significant changes after TMBIM3 expression (Figure 3g and Supplementary Figure S4g). These results suggest that the basal ER physiology and UPR signaling is not affected by TMBIM3 expression.
TMBIM3 mRNA levels are modulated by ATF4 on a mouse model of ER stress
To investigate the possible regulation of tmbim3 expression in vivo, we exposed mice to a single injection of different Tm doses, and analyzed tmbim3 mRNA levels in kidney by semiquantitative RT-PCR. A strong upregulation of tmbim3 mRNA was detected in kidney after 16 h of Tm intraperitoneal injections, which paralleled the upregulation of XBP-1 mRNA splicing and chop (Figure 3h).
Next, we investigated the possible impact of the PERK/ATF4 pathway in the transcriptional control of tmbim3 expression in vivo, and exposed atf4−/− and WT mice to Tm. We observed a significant reduction of tmbim3 upregulation in atf4−/− mice after Tm injection compared with WT control animals by semiquantitative PCR (Figure 3i). Similar results were obtained when tmbim3 mRNA levels were quantified by real-time PCR (Figure 3j). Thus, tmbim3 expression is a downstream effecter of the PERK and ATF4 signaling branch that regulates cell survival under ER stress conditions.
TMBIM3/GRINA and TMBIM3/BI-1 have complementary activities in the regulation of ER calcium homeostasis
TMBIM6/BI-1 and TMBIM4/GAAP negatively modulate ER calcium release, a phenomenon implicated in their anti-apoptotic activities.8, 16 Therefore, we monitored ER-calcium release in cells overexpressing TMBIM3 using the fluorescent Ca2+ probe Fluo-4. All experiments were performed in the nominal absence of extracellular Ca2+. Cells were treated with ATP, which leads to an intracellular inositol 1,4,5-trisphosphate (IP3) production and inositol 1,4,5-triphosphate receptor (IP3R)-mediated Ca2+ release. A significant attenuation of cytosolic Ca2+ rises was observed in TMBIM3-overexpressing cells (Figure 4a). Similar results were obtained when cells were stimulated with H2O2 (Figure 4b). Knocking down TMBIM3 in TMBIM6-deficient cells lead to a drastic increase of ER Ca2+-release after stimulation of cells with ATP (Figure 4c). Based on these results, we analyzed the susceptibility of TMBIM3-expressing cells to calcium-mediated cell death. TMBIM3 overexpression led to a significant protection against the cytotoxicity of arachidonic acid or H2O2 (Figure 4d and Supplementary Figure S5a).

TMBIM3 regulates ER calcium homeostasis and calcium-mediated cell death. MEFs cells were stably transduced with TMBIM3-MYC expression vectors and MOCK. Then, cells were loaded with the calcium indicator Fluo-4, and cytosolic calcium signals were monitored over time after stimulation in the absence of extracellular calcium with (a) 500 μM ATP, or (b) 5 mM H2O2. Left panel: a curve of calcium signal is presented representative of the analysis of at least 20 cells from three independent experiments. Right panel: the pick intensity of calcium signaling was quantified. Mean and S.E. are presented of three or four independent experiments. Student's t-test was used to analyze statistical significance (***P<0.001). (c) Alternatively, calcium signaling was monitored in TMBIM6 KO cells stably transduced with a lentiviral vector to deliver shTmbim3 or shLuc after treating them with 500 μM ATP. (d) Mock and TMBIM3-MYC-expressing cells were exposed to 200 μM araquidonic acid (Araq) or 1 mM H2O2. After 6 h of treatment, cells were stained with PI (red) and Hoechst (Blue), and visualized with a fluorescent microscope. Data is representative of three independent experiments. (e) ER-calcium content was quantified in MOCK and TMBIM3 cells after transduction of cells with adenoviruses expressing a chimeric aequorin targeted to the ER. For transient expression of TMBIM3 and its control MOCK, cells were co-transfected with expression vectors for ER aequorin. ER calcium content was monitored as described in Materials and Methods. Mean and S.E. is presented of four independent experiments. No significant (n.s.) differences Student's t-test analysis. (f) The ER 45Ca2+ content of MOCK and TMBIM3-expressing MEF cells loaded to steady state with 45Ca2+ is plotted as a function of time, depicting the passive ER 45Ca2+ leak from the ER. The figure shows a representative experiment of five independent experiments as mean and S.E.. No significant (n.s.) differences Student's t-test analysis
Previous reports indicate that TMBIM6 and TMBIM4 modulate ER calcium homeostasis by altering ER calcium content.8, 16 We quantified the ER calcium content in control and TMBIM3-overexpressing cells using the ER-targeted aequorin,17 and did not observe significant differences (Figure 4e). These results were confirmed in plasma membrane-permeabilized monolayers of control and TMBIM3-overexpressing cells using radioactive 45Ca2+ (Supplementary Figure S5b). We also measured the passive leak of Ca2+ from the ER in TMBIM3-overexpressing cells, which was unaffected upon TMBIM3 overexpression (Figure 4f). Taken together, this data suggests that TMBIM3 expression represses stimulated Ca2+ release without altering steady state-ER calcium content or Ca2+ leak.
TMBIM3/GRINA interacts with IP3R and modulates its activity
Several members of the BCL-2 family of proteins regulate ER calcium homeostasis by interacting with IP3R, either lowering steady-state ER calcium levels by sensitizing IP3R activity, or by inhibiting the Ca2+-flux properties of the IP3R, while unaffecting the ER Ca2+-store content.18, 19 We tested the possible impact of TMBIM3 expression on the direct stimulation of IP3R by the exposure of saponin-permeabilized 45Ca2+-loaded cells to increasing concentrations of IP3. TMBIM3 expression significantly reduced IP3-mediated ER calcium release, suggesting a direct regulation of the Ca2+-flux properties by TMBIM3 (Figure 5a).

TMBIM3/GRINA interacts with IP3R modulating its activity under ER stress conditions. (a) Saponin-permeabilized MOCK and TMBIM3-MYC-expressing MEFs were exposed to IP3 at indicated concentrations. The 45Ca2+ released from ER was measured, as described in Materials and Methods. Values were normalized using the Ca2+ ionophore A23187 and represent 45Ca2+ release triggered by IP3 relative to 45Ca2+ release triggered by A23187 (% of A23187-releasable 45Ca2+). Mean and S.E is presented of three to five independent experiments. Student's t-test was used to analyze statistical significance of indicated comparisons (***P<0.001). (b) TMBIM3-MYC was immunoprecipitated (IP) in MEFs cells stably expressing the protein or transduced with a MOCK expression vector and the co-precipitation of endogenous IP3R3. Results represent three independent experiments. (c) In addition, 293T cells were transfected with expression vector for TMBIM3-MYC. After 48 h, the co-precipitation of TMBIM3-MYC with endogenous IP3R3 was evaluated by western blot analysis. As control, protein expression is shown in total extracts. (d) Hela cells were transiently transfected with expression vector for TMBIM3-MYC and the co-precipitation of endogenous IP3R1 was evaluated as described in (c). (e) WT MEF cells were exposed to indicated concentrations of Tm for 16 h. Then, cells were loaded with the radiometric calcium indicator Fura-2, and cytosolic Ca2+ signals were monitored over time after ionomycin (1 μM) stimulation in the absence of extracellular Ca2+. The figure represents the mean of four independent experiments. (f) WT MEFs cells were exposed to 1 μg/ml of Tm for 16 h in presence or absence of 2-APB (10 μM) and the cell viability was evaluated by PI staining and FACS analysis. Mean and S.E. are presented of three independent experiments. Student's t-test was used to analyze statistical significance (*P<0.01). (g) MOCK and TMBIM3-MYC-expressing cells were incubated in presence or absence of Tm (100 ng/ml) for 12 h. Then, the remaining ER Ca2+ was evaluated as described in (e). The pick intensity of Ca2+ signaling was quantified. Mean and S.E are presented of four to five independent experiments. Student's t-test was used to analyze statistical significance (*P<0.001). (h) TMBIM6 KO cells stably transduced with lentiviruses expressing shLuc or shTMBIM3 construct #2 were exposed to 250 ng/ml Tm in the presence or absence of 10 μM 2-APB for 16 h. Cell viability was visualized after PI (bottom panel) and Hoechst staining (upper panel) using fluorescent microscopy (Scale bar, 60 μm). In parallel, (i) apoptotic nucleus was quantified by fluorescent microscopy. Mean and S.D. are presented of four independent experiments. Student's t-test was used to analyze statistical significance of indicated comparisons (*P<0.05)
We performed immunoprecipitation experiments to assess the possible association of TMBIM3 with IP3R-containing complexes. We immunoprecipitated the TMBIM3-MYC in stably expressing MEFs or by transient transfection in HEK cells, and observed the co-precipitation with endogenous IP3R3 (Figures 5b and c). We also observed the formation of a protein complex between TMBIM3-MYC and endogenous IP3R1 in Hela cells (Figure 5d). As control, we monitored the total expression levels of proteins involved in Ca2+ handling in TMBIM3 overexpressing and control cells, including SERCA, RYR3, and IP3R3, in addition to BCL-2, BCL-XL, and BAX (Supplementary Figure S5c).
A direct association between ER calcium release and susceptibility to ER stress has remained speculative, and only a few recent reports have provided direct molecular mechanisms between both phenomena.20, 21, 22 We tested whether ER stress alters calcium homeostasis in MEFs. Treatment of WT MEFs with Tm for 16 h led to a reduction of ER calcium content on a dose-dependent manner, as monitored by stimulation of ER calcium release with ionomycin in the absence of extracellular Ca2+(Figure 5e). In agreement with these results, treatment of cells with the IP3R inhibitor 2-aminoethoxydiphenyl borate (10 μM 2-APB) reduced ER stress-induced apoptosis (Figure 5f). We then studied the effects of TMBIM3-MYC overexpression in calcium homeostasis after exposure of cells to Tm for 12 h. TMBIM3-MYC overexpression led to a significant increase in remaining ER Ca2+ after Tm treatment (Figure 5g). Finally, the enhanced susceptibility of TMBIM6 KO cells to Tm after knocking down TMBIM3 was partially reverted after treatment of cells with 2-APB (Figures 5i and h). Thus, our results indicate that the protective effects of TMBIM3 against ER stress are because of modulation of IP3R activity.
TMBIM3/GRINA and TMBIM6/BI-1 expression have strong protective effects against ER stress in a D. melanogaster
The primary sequence of all members of the TMBIM protein family is conserved in different organisms including Drosophila melanogaster. We have employed this animal model to study UPR regulators in vivo.9, 11, 23 Using the GAL4/UAS system, we targeted UAS-RNAi transgenic flies for TMBIM3 (dTMBIM3) and TMBIM6 (dTMBIM6) using a tubulin driver coupled to GAL4. This system led to an efficient knockdown of dTmbim3 and dTmbim6 mRNA in fly larvae (Supplementary Figure S6a). We also crossed the two RNAi transgenic fly lines to produce a dTmbim3/dTmbim6 double deficient condition (doubleRNAi).
At basal level, we observed that targeting dTmbim3 or dtmbim6 with RNAi did not have a significant effect on adult fly viability (Figure 6a). Remarkably, generation of the double RNAi line leads to a significant decrease in the number of flies reaching the adult stage at basal levels (Figure 6a), suggesting a synergistic impact on fly viability. In contrast, the number of larvae reaching the pupa stage was not significantly affected (Supplementary Figures S6b and S6c).

TMBIM3 and TMBIM6 have synergistic roles in the survival against ER stress in vivo in Drosophila melanogaster. (a) D. melanogaster TMBIM3 (dTMBIM3), dTMBIM6, or both genes (double RNAi) were knocked down by expressing specific RNAi constructs using the UAS-GAL4 system under the control of a tubulin driver. Then, basal adult fly viability was investigated. Data was normalized to 100% viability with the values obtained for each RNAi control fly line. Mean and S.D. are presented for three independent experiments. Student's t-test was used to analyze statistical significance between indicated comparisons. (b) Apoptosis was monitored using second instar larvae grown in food supplemented with 25 μg/ml Tm or 0.5% DMSO (solvent control) for 24 h. Intestine of fly larvae were then collected, and active-caspase 3 was detected by immunofluorescence (green). Nucleus was stained with DAPI (blue) to visualize total number of cells. Images represent the analysis of three independent experiments. (c) Life cycle was monitored using second instar larvae grown in food supplemented with 25 μg/ml Tm dissolved in DMSO or 0.5% DMSO as control in dTMBIM3, (d) dTMBIM6, or (e) both genes (double RNAi) were knocked down with RNAi. The number of individuals reaching the adult fly stage was evaluated over time starting from 100 viable larvae. Left panel: a representative experiment is presented following the survival of 100 larvae individuals over time. Right panel: quantification of the total adult flies obtained in a representative experiment after 15 days. Mean and S.E. are presented of three independent experiments. Student's t-test was used to analyze statistical significance (*P<0.05; **P<0.01; ***P<0.001)
We then exposed fly larvae to 25 μg/ml Tm or control solvent (DMSO) in the growing media, and then monitored the rate of apoptosis induction in the intestine of the larvae. Larvae intestines were dissected and stained with an anti-active caspase-3 (ICE) antibody together with Hoechst to visualize the total number of cells. As shown in Figure 6b, knocking down dTmbim3 or dTmbim6 leads to an increased basal apoptosis, which was further enhanced when larvae were exposed to Tm.
We also determined the functional impact of dTmbim3 and dTmbim6 expression in animal survival after exposure to the ER stress agent Tm by measuring the number of individuals that reached the pupa stage or matured into adult flies. Exposure of fly larvae to Tm leads to a drastic increase in the toxicity when dTmbim3 or dTmbim6 (recently reported in Castillo et al.11) were knocked down, observing a significant reduction in the number of adult flies of 38% and 43%, respectively, when compared with untreated larvae (Figures 6c and d). Remarkably, double RNAi flies for dTmbim3 and dTmbim6 were highly susceptible to the exposure to Tm, detecting minimal survival of adult flies (Figure 6e). Taken together, these experiments suggest that TMBIM3 and TMBIM6 have anti-apoptotic activities in vivo in D. melanogaster.
TMBIM3 deficiency increases the rate of apoptosis during development in zebrafish
Bioinformatic analysis of zebrafish genomic databases indicated the presence of a putative TMBIM3 homolog (Supplementary Figure S1a, zebrafish tmbim3 (zTmbim3)). We confirmed the expression of the predicted zTmbim3 transcript in embryos (Supplementary Figure S7a). Using in situ hybridization, we detected a marked expression of zTmbim3 mRNA in neuronal tissue, including head, eye, and spinal cord tissue (Figure 7a). Consistent with these results, analysis of mouse TMBIM3 (mTmbim3) mRNA levels in different mouse tissues by real-time PCR also indicated a significant expression in spinal cord, cerebellum, brain, in addition to kidney and testis (Supplementary Figure S7b).

Expression pattern and impact of knocking down TMBIM3 in zebrafish. (a) Pattern of expression of zTmbim3 mRNA as visualized by whole mount in situ hybridization at three developmental stages: 8 cells (i), 10 somites (ii) and 24 hpf (iii–v). Images correspond to lateral (i–ii) and dorsal (iv–v) views of embryos. (b) Effect of zTmbim3 knock down on cell death during zebrafish development. Acridine orange staining was assessed in 24 hpf embryos after injection of 1.5 ng of either control mismatch morpholino (Mis-MO) or zTmbim3-MO. Images of bright field (left panel) and fluorescence (right panel) correspond to lateral views of embryos, and are representative of three independent experiments. (c) Quantification of acridine orange staining (pixel intensity/area) in the spinal cord of zTmbim3-depleted embryos (dashed square depicted in panel (b)). Data represent mean and S.D. of the analysis of 20 embryos of three independent experiments. Student's t-test was used to analyze statistical significance (**P<0.01). (d) Quantification of activated caspase-3 fluorescent immunoreactivity in the spinal cord of zTmbim3-depleted embryos as described in (c). Data represent mean and S.D. of the analysis of three embryos. Student's t-test was used to analyze statistical significance (*P<0.05). (e) Panels show dorsal views of whole-mounted embryos, oriented anterior to the left (A) and posterior to the right (P). Zebrafish Tg(ngn1∷GFP) embryos were injected with 4.5 ng of either Mis-MO in one-cell stage. Embryos were then visualized in vivo at 24 hpf by confocal microscopy. Data represents the analysis of 20 embryos for each condition. GFP-labeled telencephalon (t), eye (e), third ventricle (3e), dorsal midbrain (m), interneurons (in) Rohon Beard sensory neurons (rb) over the dorsal longitudinal fasciculus (dlf). The boundaries of the spinal cord are delineated with a red arrow of felt double. Scale bars represent 20 μm. (f) One-cell stage zebrafish embryos were injected with 4.5 ng of either Mis-MO or zTmbim3-MO, in the presence or absence of 50–100 μg of either control (Gap43-cherry) or mTmbim3 mRNAs. Embryos were then visualized with bright field at 60–70% epiboly or 24 hpf. Data represents the analysis of 346 embryos. (g) Quantification of the percentage of surviving embryos at 24 hpf treated in (f). Data represent the average of two independent experiments (n=127 and 219 embryos analyzed per experiment, respectively)
In order to knockdown zTmbim3, we designed a morpholino antisense oligonucleotide against the 5′-UTR and ATG region of zTmbim3, (Supplementary Figure S7c). We then injected 1.5 ng per embryo of zTmbim3 morpholino (zTmbim3-MO) or a mismatch control morpholino into zebrafish-fertilized ovocytes. After 24 h post-injection, knocking down zTmbim3 drastically increased the amount of dying cells, which were enriched in the tail and spinal cord (Figures 7b–d). Interestingly, higher doses of zTmbim3-MO (4.5 ng per embryo) led to massive neuronal loss in the brain, reflected in evident morphological changes (Supplementary Figure S7d). To corroborate this finding, we injected zTmbim3-MO in a transgenic zebrafish specifically expressing neuronal green fluorescent protein (GFP) to visualize cell morphology and viability. GFP-expressing embryos demonstrated massive neural death in brain and spinal cord, along with disorganization of the neural circuits (Figure 7e). In contrast, this treatment did not affect gastrulation as monitored in embryos at 70% epiboly (Figure 6f), suggesting that early developmental processes are not dependent on zTmbim3 expression. The phenotype was fully rescued by overexpressing mTmbim3 through microinjecting in vitro-transcribed mRNA (Figures 7f and g).
TMBIM3 protects zebrafish embryos against ER-related injuries
We developed a model to induce ER stress in zebrafish by treating embryos with Thg at 24 h post fertilization (hpf) (Figures 8a and b). Remarkably, an induction of zTmbim3 was observed in zebrafish embryos after 4 h of Thg treatment, as monitored by real-time PCR (Figure 8b). To investigate the effect of TMBIM3 expression in ER stress-induced cell death, we overexpressed the protein by microinjecting mTmbim3 mRNA into zebrafish embryos at one-cell stage (Figure 8c). Then, embryos at 8 hpf were treated with Thg for 4 h, and cell death monitored by acridine orange staining. As shown in Figure 8d, mTmbim3 overexpression led to a significant protection against Thg in vivo. Interestingly, we notice that treatment of zebrafish embryos at 24 phf with Thg for 4 h lead to an evident curvature and atrophy of the tail, a phenotype fully reverted by the overexpression of mTmbim3 (Figure 8e). Taken together, our results indicate an essential role of zTmbim3 in apoptosis during development and in the regulation of ER stress responses in vivo.

TMBIM3 protects against ER stress in zebrafish in vivo. (a) Embryos at 24 hpf were exposed to 1 and 5 μM Thg for 4 h, and then zebrafish xbp-1 mRNA splicing was evaluated by RT-PCR. PCR fragments corresponding to spliced and unspliced forms are indicated. Actin levels were monitored as loading control. (b) In embryos described in (a), the mRNA levels of the zebrafish chop (zChop) and zTmbim3 were measured by real-time PCR, and normalized by actin expression. Data represents mean of the analysis of two independent experiments performed in triplicates. (c) One-cell stage embryos were injected with 50–100 pg of in vitro transcribed mouse tmbim3 (mTmbim3) mRNA. After 24 h, mRNA levels of the exogenous mTmbim3, and endogenous zTmbim3 were evaluated by RT-PCR as control for experiments presented in (d). (d) Cell death was monitored in 8 hpf embryos after exposition to indicated concentrations of Thg for 4 h. Then, embryos were stained with acridine orange. Insets show the corresponding bright field images of the embryos for each condition. Data represents three independent experiments of a total 3–5 embryos analyzed per determination. (e) Changes in tail morphology were monitored at 24 hpf after Thg treatments as described in (d). Arrows indicate alterations in tail morphology. Data is representative of three independent experiments of a total 10–15 embryos visualized per experiment. (f) Working model. Overall, our data suggest that TMBIM3 is a highly conserved anti-apoptotic protein that regulates cell survival under ER stress conditions. Activation of the PERK/ATF4 signaling branch of the UPR leads to the upregulation of tmbim3 mRNA under ER stress conditions. TMBIM3 negatively modulates the release of ER calcium by IP3R, decreasing the sensibility of cells to ER stress and calcium-mediated cell death. TMBIM3 has synergistic effects with TMBIM6, which may be associated with the formation of a protein complex containing TMBIM6
Discussion
Historically, most of the attention in the field of apoptosis regulation in mammals has been focused on addressing the role of the BCL-2 family of proteins in the process at the mitochondria. This is based, in part, on seminal findings identifying BCL-2 as the cause of certain forms of B cell leukemia in humans, and its subcellular location at the mitochondria. Although BCL-2-related proteins are essential in the regulation of cell death in mammalian models, most members are poorly conserved in other species where regulated cell death has an active role in development and adult tissue physiology.1, 24 In contrast to the BCL-2 family, genetic and bioinformatics studies revealed that TMBIM family members are highly conserved in evolution,6, 7 with a possible common ancestor in yeast (Supplementary Figure S1c).25
Only a few studies have addressed the possible function of other TMBIM family members in cellular models, with poor mechanistic insights. Moreover, possible functional interactions between TMBIM family members have not been explored so far. TMBIM2/LFG has a plasma membrane location, and attenuates Fas ligand-induced apoptosis.26 TMBIM1/RECS1 has a lysosomal, Golgi, and plasma membrane location,27, 28 and only one study addressed its impact on apoptosis, where it interacts and inhibit Fas ligand-mediated apoptosis.29 TMBIM4/GAAP is exclusively located at the Golgi compartment and its expression modulates the susceptibility of cells to intrinsic and extrinsic apoptotic stimuli.16, 30 TMBIM5/GHTIM is located at the mitochondria, and has been shown to prevent mitochondrial fragmentation and the release of cytochrome c induced by actinomycin D treatment.31 Finally, a recent screening to identify regulators of Shiga toxin-induced cell death found an artificial truncated fragment of TMBIM3/GRINA cDNA as a possible inhibitor of its toxicity.32
Most studies addressing the role TMBIM family members in vivo have been focused on TMBIM6. TMBIM6 is implicated in disease condition including cancer,6 ischemia-reperfusion,8 diabetes and obesity, bacterial infections, and liver regeneration.5, 6 TMBIM2 knock-out mice are also viable, and depict an enhanced susceptibility to cystic medium degeneration and aortic dilation.27 A putative TMBIM4 homolog is present in Vaccinia virus, and its overexpression affected virus virulence in a murine infection model.16 An early report two decades ago identified a putative TMBIM3/GRINA homolog/ortholog in Rattus sp neurons (NMDARA1; also called glutamate-binding protein.33 However, further studies led to doubts about its identity as a glutamate receptor.34 Interestingly, human TMBIM3 gene location maps close to a locus genetically linked with epilepsy.35 A viable tmbim3 knock-out mice was recently generated confirming the neuronal expression of TMBIM3 in mice.36 However, no functional analysis of TMBIM3 KO mice was performed.
Here we have investigated the possible impact of TMBIM3 expression on the regulation of apoptosis. Gain and loss of functional studies revealed a highly specific role of TMBIM3 in the control of ER stress-mediated apoptosis. Remarkably, both in vitro and in vivo approaches in a D. melanogaster revealed that TMBIM3 and TMBIM6 have a potent and synergistic pro-survival activity. This effect on cell death/animal survival contrasts with several studies showing poor apoptosis-related phenotypes of BCL-2/buffy or BAX/debcl mutants in D. melanogaster.3, 4 We also investigated the function of TMBIM3 in apoptosis using a vertebrate model. Targeting TMBIM3 expression in zebrafish demonstrated enhanced apoptosis during development, and drastic alterations in brain morphology and neuronal survival, in addition to protective effects on an in vivo paradigm of ER stress. Our results suggest that TMBIM3 and TMBIM6 have complementary activities in the control of Ca2+ signaling and ER stress-mediated apoptosis (model in Figure 8f), reflected in the appearance of striking phenotypes when both proteins were manipulated at the same time. These observations suggest that a complex regulatory network may operate at the ER membrane, where several TMBIM family members functionally interact and synergize in the control of cell death. Our current study exemplifies the need for a systematic analysis to address the role of TMBIM family of proteins in apoptosis, and to further define the hierarchical organization between their components.
Materials and Methods
Reagents
Tm, Thg, staurosporine, etoposido, actinomicin D, and TNFα were purchased from Calbiochem EMD Bioscience Inc (Darmstadt, Germany). Cell culture media, fetal calf serum, and antibiotics were obtained from Life Technologies (Grand Island, NY, USA). Hoechst, Brefeldin A-BODIPY, and ALEXA secondary antibodies were purchased from Molecular Probes (Grand Island, NY, USA). Anti-IgD and anti-IgM fluorescent antibodies were purchased from BD Biosciences (Baltimore, MA, USA).
Cell culture and DNA constructs
Human BI-1 was cloned into pCDNA.3 and MYC-tagged. Mouse TMBIM3 was cloned from a cDNA library and cloned into pCR3.1 vector and MYC-tagged. Then, cDNAs were subcloned into pMSCV-puro retroviral vector (Clontech, Mountain View, CA, USA). PERK- and IRE1α-deficient MEFs cells were obtained from David Ron, and ATF6α KO cells from Dr. Randal Kaufman. We generated transient and stable MEFs with reduced levels of TMBIM3 mRNA using methods previously described,9 with shRNA using the lentiviral expression vector pLKO.1. As control, an shRNA construct against the luciferase gene was employed.9 shRNA constructs were generated by the Broad Institute (Boston, MA, USA) based on different criteria for shRNA design (http://www.broad.mit.edu/genome_bio/trc/rnai.html). We screened a total of five different constructs and selected the two most efficient construct after real-time PCR analysis (shRNA#1 and shRNA#2) corresponding to the target sequence 5′-GCTGCATTTCTGTGCCACCTT-3′ and ‘5’-CTGGACCATCATTGTCTCCTA-3′. ATF6 shRNA were described before.37
Viability assay
In all, 2.0 × 104 cells were seeded in 24-well plate and the maintained by 24 h in DMEM cell culture media supplemented with 5% bovine fetal serum and non-essential amino acids. ER stress was induced by adding ER stress agents to the cells at different concentrations, and maintained for 24 h. To induce nutrient starvation, cell culture media was replaced by Earle's balanced salt solution or RPMI without glucose and serum, and incubated for 36, 24, 18, 12 or 6 h. Then, cell viability was monitored as previously described using propidium iodide staining and flow cytometry (BD FACS Canto, Biosciences). In parallel, nuclear morphology was analyzed by Hoechst333342 staining (Molecular Probes). For cell re-plating experiments, 3.0 × 105 cells were harvested in 3.5 cm dishes and treated with 1 μg/ml of Tm for 4 h, or exposed to Earle's balanced salt solution or RPMI for 6 h. Cells were washed and then trypsinized. A total of 2 × 105 MEFs cells were re-plated into 10 cm dishes and cultured for 5 days in regular DMEM media supplemented with 5% serum and non-essentials amino acids. At 5 days after treatment, the cells were washed three times in PBS, fixed with 4% PFA, stained with crystal violet for 30 min, and then washed three times with water.
Western blot analysis
Cells were collected and homogenized in RIPA buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.5% DOC, and 0.5% Triton X-100) containing a protease inhibitor cocktail (Roche, Basel, Switzerland) by sonication. Protein concentration was determined by micro-BCA assay (Pierce, Rockford, IL, USA). The equivalent of 15–50 mg of total protein was loaded onto 4–12%, 7.5%, 12%, or SDS-PAGE minigels (Cambrex, East Rutherford, NJ, USA) depending on the analysis. The following antibodies and dilutions were used: anti-XBP-1 1 : 1000, anti-HSP90 1 : 4000, anti-ATF4 1 : 2000, anti-CHOP 1 : 2000, anti-SP-1 1 : 1000, anti-BAX 1 : 2000, anti-myc 1 : 1000, anti BCL-2 1 : 5000 (Santa Cruz, Santa Cruz, CA, USA), anti-BAK 1 : 2000 (Upstate Technology, Billerica, MA, USA), and anti-P-eIF2α 1 : 2000, anti-PERK 1 : 2000, anti-active caspase-3 (Cell Signaling Technology, Berverly, MA, USA), anti-PDI, anti-ERp57, anti-BiP 1 : 2000 (StressGen, Enzo Life Sciences, Farmingdale, NY, USA), anti-SERCA (Affinity Bioreagents, Rockford, IL, USA), and anti-IP3-R3 1 : 5000 (BD Biosciences).
RNA extraction and RT-PCR
Total RNA was prepared from cells and tissue using Trizol (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesized with SuperScript III (Invitrogen) using random primers p(dN)6 (Roche). Quantitative real-time PCR reactions employing SYBRgreen fluorescent reagent were performed in an ABI PRISM7700 system (Applied Biosystems, Foster City, CA, USA). The relative amounts of mRNAs were calculated from the values of comparative threshold cycle by using b-actin as control. Primer sequences were designed by Primer Express software (Applied Biosystems) or obtained from the Primer Data bank (http://pga.mgh.harvard.edu/primerbank/). Most real-time PCR were previously described.9 In addition, we used the following primers: mouse TMBIM3 5′-CCCTACCCTCAAGGAGGCTAC-3′ and 5′-CTGGCGAATGTTCTTGTCCC-3′, D. melanogaster TMBIM3 5′-GCTTAAGTTGAGGCGCAAAC-3′ and 5′TTGAAAATCGGGATTCCTTG-3′, zebra flish TMBIM3 5′-ATTGTTGGAGTGGATTATGA-3′ and 5′-CAGGTTGTTAATCTGAACAT-3′, zebra fish CHOP 5′-ACCCTGAATCAGAAGCAGCCG-3′ and 5′-TACGACACGCTCCCACTCCT, zebra fish actin 5′-ACACGACCCAGAGCATCAGGGAG and 5′-CCTCTCTTGCTCTGAGCCTCA-3′. For zebra fish splicing, the following primers were used: 5′-GTTCAGGTACTGGAGTCCGA-3′ and 5′-CTCAGAGTCTGCAGGGCCAG-3′.
Cloning of zebrafish (Danio rerio) Tmbim3
The 5′UTR and part of the coding region of zebrafish tmbim3 (zTmbim3) were amplified by PCR using cDNA from 24 hpf embryos. The following primers were design from a known zebrafish Tmbim3 sequence (NM_201208): forward 5′-GGGATGTTTACAGGAAGACGAG-3′ and reverse 5′-ACAAACGCAGTGGTGATGG-3′. The PCR product was cloned in pCRII-TOPO vector (Invitrogen) (pCRII-TOPO-Dr-tmbim3) and sequenced.
Whole-mount in situ hybridization and immunofluorescence of zebrafish embryos
A DIG-labeled antisense RNA probe for Dr-tmbim3 was synthesized with T7 RNA polymerase and DIG-labeled UTP (Roche) using linearized pCRII-TOPO-Dr-tmbim3 as a template. Whole-mount in situ hybridization was performed according to standard protocols for zebrafish. After staining, labeled embryos were mounted in 100% glycerol and examined on a Nikon (Melville, NY, USA), DIC microscope (Eclipse 80i). Caspase-3 assay was performed in embryos fixed in 4% paraformaldehyde overnight at 4°C and then transferred to 100% methanol overnight at 4°C. Anti-activated human Caspase-3 antibody (Cell signaling, 1 : 200) and Alexa-488 rabbit conjugated secondary (Molecular Probes, 1 : 200) antibodies were used. Incubation with To-Pro-3 iodidestain (642/661) (Molecular Probes, 1 : 1000) for 1 h was used for nuclear counterstaining. Embryos were mounted in glycerol and evaluated by confocal microscopy LSM-140 Axiovert 10.0, Zeiss (Thornwood, NY, USA). Images were processed using ImageJ software (NIH, Boston, MS, USA).
Morpholino knock down in zebrafish
Morpholino antisense oligonucleotides (MO) were designed and synthesized by Gene Tools (Philomath, OR, USA). Sequences were as follows: 5′-GCCTGTATCCCTTATTCTGAGACAT-3′ (zTmbim3-MO) and 5′-GCgTcTATCgCTTATTgTcAGACAT-3′ (mismatch control morpholino). Lower case letters indicate the five base pair mismatch in control MO. Pressure injection of antisense morpholino (1.5–4.5 ng per embryo) was performed in one-cell stage zebrafish embryos using standard protocols WT Tübingen, Tg(HuC∷GFP)zebrafish embryos using standard protocols.
Synthesis of RNA and rescue assay in zebrafish
Synthetic capped RNA was generated with the T7 mMESSAGE mMACHINE system (Ambion, Foster City, CA, USA) using aXhoI-linearized version of the mouse TMBIM3-pCT3.1 vector as a template. Sense mRNA (50 pg per embryo) was microinjected in one-cell stage zebrafish embryos. For rescue experiments, zebrafish embryos were co-injected at one-cell stage with a combination of 50 pg mTmbim3 mRNA+4.5 ng zTmbim3-MO, or 50 pg gap43-Cherry-pCS2 mRNAs+4.5 ng zTmbim3-MO (or mismatch-MO). Phenotypes were analyzed at 60–70% epiboly and 24 hpf.
In vivo cell death assay in zebrafish
For in vivo detection of apoptotic cells, WT Tübingen embryos were injected at 1-cell stage with 1.5 ng z-tmbim3-MO or mismatch-MO, and maintained for 24 h in E3 medium at 28.5°C. After dechorionation, embryos were incubated with E3 medium containing 2 mg/ml acridine orange for 30 min at 28, 5°C, using 24-well plates (6 embryos per well). After eight 5 min washes with E3 medium, embryos were anesthetized with tricaine (Sigma-Aldrich, St. Louis, MO, USA) and then results were documented by fluorescence microscopy. All fluorescent pictures were taken at exactly the same exposure, gain, and magnification. The region of interest analysis was performed in the spinal cord region and the quantification of fluorescence was performed with ImageJ software (NIH, USA). In order to demonstrate loss of brain and spinal cord neurons, transgenic embryos Tg(ngn1∷GFP) were injected with 4.5 ng per embryo of z-tmbim3-MO or mismatch control-MO and incubated for 24 h at 28.5°C. In vivo embryos were mounted in agarose and analyzed by confocal microscopy LSM-140 Axiovert 10.0, Zeiss. Images were processed using ImageJ software (NIH, USA) and Volocity (Improvisión, Coventry, England).
To evaluate the protective effect of tmbim3 in ER stress, zebrafish embryos were first injected with 50–100 pg mTmbim3 or gap43-Cherry-pCS2 (control) mRNAs. Then, after 8 and 24 h, embryos were incubated for 4 h with E3 medium containing 1–5 μM Thg at 28.5°C, using 24-well plates (6 embryos per well). Finally, apoptotic cells were evaluated in vivo by staining with acridine orange (8 hpf) or through assessment of morphological changes (24 hpf).
Drosophila experiments
Flies were kept at 25°C on standard medium with a 12–12 dark-light cycle. The line Tub-Gal4/TM3-GFP was obtained from the Bloomington Drosophila Stock Center (Bloomington, In, USA). UAS-RNAi lines were obtained from the Vienna Drosophila RNAi Center: UAS-TMBIM3RNAi(#28365) and UAS-BI-1RNAi.38 For the RNAi experiments, female UAS-RNAi were crossed to male Tub-Gal4/TM3-GFP flies. Second instar GFP-positive larvae (not expressing the RNAi) were separated from GFP-negative larvae (expressing the RNAi) and fed separately with Tm or the vehicle (DMSO). In survival experiments, an indicated number of second instar larvae of the indicated genotype were fed with 25 μg/ml Tm or DMSO in instant Drosophila food (Carolina Biological Supply 2700 York Burlington, NC, USA). The total number of pupae, adult flies eclosed and flies alive after 5, 10 and 15 days was counted and the percentages from the total larvae were determined. For immunohistochemistry, third instar larval intestines were dissected and stained using standard procedures. Briefly, the tissue was fixed with 4% paraformaldehyde in PBS with 0.3% triton for 30 min. Then, the tissue was blocked in PBS-0.3% triton and 0.5% goat serum for 1 h. The tissue was incubated with slow rotation at 4°C overnight with rabbit anti-caspase3 (Cell Signaling) antibody (1 : 300). After washing, the secondary antibody anti-rabbit conjugated with fluorescein (1 : 200, Jackson ImmunoResearch, West Grove, PA, USA) was added and incubated for 1–2 h at room temperature. The tissue was mounted with DAPI in vectashield (Vector Laboratories, Burlingame, CA, USA) and observed in a spinning disc microscopy.
Calcium measurements
Cells were grown in coverslips and then loaded with Fluo-4 AM (1 μM) or Fura-2 (1 μM) for 30 min at room temperature. Then coverslips were mounted in a 1-ml capacity chamber and washed three times with Ca2+-free buffer (150 mMNaCl, 5 mMKCl, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose and 5 m MEGTA, pH 7.4). Ca2+-free buffer was perfused 2 min before set-off Ca2+ release from intracellular stores. Then, drugs (ionomycin, ATP, Thg, and H2O2) were applied in the nominal absenceof Ca2+. Ca2+signals were recorded using an IX-81-inverted microscope for fluorescence measurements (DSU, Olympus, Tokyo, Japan), equipped with a 150-W xenon lamp (Olympus MT-20). Fluo-4 fluorescence was excited and detected with a FITC filter cube, using a 40 × /1.4 NA oil immersion objective. Fura-2 was alternately excited at 340 and 400 nm, and the fluorescence filtered at 510 nm, collected and recorded at 0.2 Hz using a CCD-based imaging system (Olympus, DSU) running CellR software (Olympus). Changes in cytosolic calcium were measured in a field-of-view consisting of 15–30 cells. Images were acquired every 5 s. The mean intensities of small cellular areas of interest were collected as F(t) and the background intensity was subtracted, using a same-size region of interest outside the cell, yielding F(t)s. In Fluo-3 assays, the final signal was normalized to baseline fluorescence F(0), as (F(t)s−F(0))/F(0). In Fura-2 assays, results are expressed as the ratio between the 340 nm and 400 nm (R340/400) signals over the baseline.
ER calcium content measurements and unidirectional 45Ca2+-flux assays
For luminescence measurement of Ca2+, 5 × 104 WT MEFs cells were seeded on 13-mm glass coverslips. After 24 h, the cells were co-transfected with 0.2 μg of ER-aequorin39 and 0.8 μg of TMBIM3-MYC or empty vector (MOCK) using Lipofectamine LTX (Invitrogen). For stable cell line, MOCK and TMBIM3 cells were transduced with Ad-ER-aequorin.
These experiments were performed on WT MEF cells and TMBIM3-MYC overexpressing MEFs grown to confluent monolayers, as described before (Rong et al., Mol Cell, 2008; Decuypere et al., Cell Calcium 2010). Cells were grown in 12-well plates for 5–6 days after seeding at 20 000 cells per well. The wells were fixed on a thermostated plate at 25 °C on a mechanical shaker. The culture medium was aspirated, and the cells were permeabilized by incubating them for 10 min in a solution containing 120 M MKCl, 30 mM imidazole-HCl (pH 6.8), 2 mM MgCl2, 1 mM ATP, 1 mM EGTA and 20 μg/ml saponin. The non-mitochondrial Ca2+ stores were then loaded for 45 min in 120 mM KCl, 30 mM imidazole-HCl (pH 6.8), 5 mM MgCl2, 5 mM ATP, 0.44 mM EGTA, 10 mM NaN3 and 150 nM free 45Ca2+ (28 μCi/ml). Then, cells were washed twice with 1 ml of efflux medium containing 120 mM KCl, 30 mM imidazole-HCl (pH 6.8) and 1 mM EGTA supplemented 4 μM Thg to block SERCA Ca2+-uptake activity. Then, 500 μl of efflux medium was added and replaced every 2 min. After 10 min of efflux, five different [IP3] (wells 1–10) and 10 μM of Ca2+ionophore A23187 (wells 11 and 12) were applied for 2 min. Eight minutes later, all 45Ca2+ remaining in the stores was released by incubation with 1 ml of a 2% (w/v) sodium dodecyl sulfate solution for 30 min. Differences between concentration-response curves were analyzed using two-way ANOVA. Experiments to determine the ER 45Ca2+-leak rate were performed in similar ways, except that IP3 was omitted during the efflux and that one condition was included in which the cells were loaded in the presence of 10 μM A23187 in order to determine the non-specific binding of 45Ca2+. The specific ER 45Ca2+-leak rate was determined by subtracting the 45Ca2+ content in the presence of 10 μM A23187 from the total 45Ca2+ content.
Immunoprecipitation
All immunoprecipitations were performed using CHAPS buffer as described in Lisbona F et al.9
Abbreviations
- 2-APB:
-
2-aminoethoxydiphenyl borate
- ATF4:
-
activating transcription factor 4
- ATF6:
-
activating transcription factor 6
- BI-1:
-
bax inhibitor 1 protein
- ERAD:
-
endoplasmic reticulum-associated degradation
- Est:
-
estaurosporine
- Eto:
-
etoposide
- GAAP:
-
golgi anti-apoptotic proteín
- GFP:
-
green fluorescent protein
- GHITM:
-
growth-hormone inducible transmembrane protein
- GRINA:
-
glutamate receptor, ionotropic, N-methyl D-asparate-associated protein 1
- HA:
-
human influenza hemagglutinin
- hpf:
-
hours post fertilization
- IP3:
-
inositol 1,4,5-trisphosphate
- IP3-R:
-
inositol 1,4,5-triphosphate receptor
- Mo:
-
morpholine
- shRNA:
-
small hairpin RNA
- Thg:
-
thapsigargin
- Tm:
-
tunicamycin
- TMBIM:
-
transmembrane BAX inhibitor motif-containing
- UPR:
-
unfolded protein response
References
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Hengartner MO . Programmed cell death in invertebrates. Curr Opin Genet Dev 1996; 6: 34–38.
Sevrioukov EA, Burr J, Huang EW, Assi HH, Monserrate JP, Purves DC et al. Drosophila Bcl-2 proteins participate in stress-induced apoptosis, but are not required for normal development. Genesis 2007; 45: 184–193.
Galindo KA, Lu WJ, Park JH, Abrams JM . The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development 2009; 136: 275–283.
Robinson KS, Clements A, Williams AC, Berger CN, Frankel G . Bax inhibitor 1 in apoptosis and disease. Oncogene 2011; 30: 2391–2400.
Reimers K, Choi CY, Bucan V, Vogt PM . The Bax Inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med 2008; 8: 148–156.
Hu L, Smith TF, Goldberger G . LFG: a candidate apoptosis regulatory gene family. Apoptosis 2009; 14: 1255–1265.
Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell 2004; 15: 355–366.
Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 2009; 33: 679–691.
Xu Q, Reed JC . Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol Cell 1998; 1: 337–346.
Castillo K, Rojas-Rivera D, Lisbona F, Caballero B, Nassif M, Court FA et al. BAX inhibitor-1 regulates autophagy by controlling the IRE1alpha branch of the unfolded protein response. EMBO J 2011; 30: 4465–4478.
Hetz C, Martinon F, Rodriguez D, Glimcher LH . The unfolded protein response: integrating stress signals through the stress sensor IRE1{alpha}. Physiol Rev 2011; 91: 1219–1243.
Hetz C, Glimcher LH . Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell 2009; 35: 551–561.
Woehlbier U, Hetz C . Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci 2011; 36: 329–337.
Asada R, Kanemoto S, Kondo S, Saito A, Imaizumi K . The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. J Biochem 2011; 149: 507–518.
Gubser C, Bergamaschi D, Hollinshead M, Lu X, van Kuppeveld FJ, Smith GL . A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog 2007; 3: e17.
Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T et al. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 2000; 148: 857–862.
Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A 2009; 106: 14397–14402.
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A 2005; 102: 105–110.
Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M et al. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 2010; 68: 865–878.
Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR et al. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol 2009; 186: 783–792.
Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008; 27: 285–299.
Mori K . Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 2009; 146: 743–750.
Metzstein MM, Hengartner MO, Tsung N, Ellis RE, Horvitz HR . Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans gene ces-2. Nature 1996; 382: 545–547.
Huckelhoven R . BAX Inhibitor-1, an ancient cell death suppressor in animals and plants with prokaryotic relatives. Apoptosis 2004; 9: 299–307.
Somia NV, Schmitt MJ, Vetter DE, Van Antwerp D, Heinemann SF, Verma IM . LFG: an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci U S A 1999; 96: 12667–12672.
Zhao H, Ito A, Kimura SH, Yabuta N, Sakai N, Ikawa M et al. RECS1 deficiency in mice induces susceptibility to cystic medial degeneration. Genes Genet Syst 2006; 81: 41–50.
Zhao H, Ito A, Sakai N, Matsuzawa Y, Yamashita S, Nojima H . RECS1 is a negative regulator of matrix metalloproteinase-9 production and aged RECS1 knockout mice are prone to aortic dilation. Circ J 2006; 70: 615–624.
Shukla S, Fujita K, Xiao Q, Liao Z, Garfield S, Srinivasula SM . A shear stress responsive gene product PP1201 protects against Fas-mediated apoptosis by reducing Fas expression on the cell surface. Apoptosis 2011; 16: 162–173.
de Mattia F, Gubser C, van Dommelen MM, Visch HJ, Distelmaier F, Postigo A et al. Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Mol Biol Cell 2009; 20: 3638–3645.
Oka T, Sayano T, Tamai S, Yokota S, Kato H, Fujii G et al. Identification of a novel protein MICS1 that is involved in maintenance of mitochondrial morphology and apoptotic release of cytochrome c. Mol Biol Cell 2008; 19: 2597–2608.
Yamaji T, Nishikawa K, Hanada K . Transmembrane BAX inhibitor motif containing (TMBIM) family proteins perturbs a trans-Golgi network enzyme, Gb3 synthase, and reduces Gb3 biosynthesis. J Biol Chem 2010; 285: 35505–35518.
Kumar KN, Tilakaratne N, Johnson PS, Allen AE, Michaelis EK . Cloning of cDNA for the glutamate-binding subunit of an NMDA receptor complex. Nature 1991; 354: 70–73.
Hollmann M, Heinemann S . Cloned glutamate receptors. Annu Rev Neurosci 1994; 17: 31–108.
Bonaglia MC, Giorda R, Tenconi R, Pessina M, Pramparo T, Borgatti R et al. A 2.3 Mb duplication of chromosome 8q24.3 associated with severe mental retardation and epilepsy detected by standard karyotype. Eur J Hum Genet 2005; 13: 586–591.
Nielsen JA, Chambers MA, Romm E, Lee LY, Berndt JA, Hudson LD . Mouse transmembrane BAX inhibitor Motif 3 (Tmbim3) encodes a 38 kDa transmembrane protein expressed in the central nervous system. Mol Cell Biochem 2011; 357: 73–81.
Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009; 23: 2294–2306.
Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007; 448: 151–156.
Pinton P, Brini M, Bastianutto C, Tuft RA, Pozzan T, Rizzuto R . New light on mitochondrial calcium. Biofactors 1998; 8: 243–253.
Acknowledgements
We apologize to all colleagues whose work could not be cited owing to space limitations. We thank David Ron for providing IRE1α and PERK-deficient cells, Dr. Ann-Hwee Lee for providing and generating IRE1α KO-reconstituted cells, and Dr. NirHacohen and the Broad Institute (Boston, MA, USA) for providing shRNA lentiviral constructs. We also thank Dr. John Reed for kindly providing bi-1−/−cells and Dr. Randal Kaufman for providing ATF6α-deficient cells. We thank Peter Thielen for technical assistant in cloning. We thank Cecilia Zuñigafor for his technical support with fluorescence-activated cell sorting analysis, Tomas Luyten for technical support with 45Ca2+-flux assays and Jan Parys for helpful discussions. This work was supported by the FONDECYT no. 1100176, Alzheimer's Association, Muscular Dystrophy Association, Michael J Fox Foundation for Parkinson Research, North American Spine Society, and ICGEB (to CH); FONDECYT no.1090272 (JS), FONDECYT no. 11090324(AC), Howard Hughes Medical Institute (INTNL 55005940), FONDECYT (1090242) and the Millennium Initiative (ICM P07-048-F) (MC); FONDECYT no. 3100033 (DR); CONICYT Doctoral fellowship (DR-R, GM); UCH-0606 (DR-R); CONICYT no. 24090143 (DR-R), Millennium Nucleus no. P07-048-F (CH, MC, JS); FONDAP Grant no. 15010006 (RA, AS and CH); Grants STRT1/10/044 (GB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H Ichijo
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Rojas-Rivera, D., Armisén, R., Colombo, A. et al. TMBIM3/GRINA is a novel unfolded protein response (UPR) target gene that controls apoptosis through the modulation of ER calcium homeostasis. Cell Death Differ 19, 1013–1026 (2012). https://doi.org/10.1038/cdd.2011.189
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.189
Keywords
This article is cited by
-
miR-125a-3p regulates apoptosis by suppressing TMBIM4 in lens epithelial cells
International Ophthalmology (2022)
-
Mechanisms, regulation and functions of the unfolded protein response
Nature Reviews Molecular Cell Biology (2020)
-
Genotoxic stress triggers the activation of IRE1α-dependent RNA decay to modulate the DNA damage response
Nature Communications (2020)
-
PRKCSH contributes to tumorigenesis by selective boosting of IRE1 signaling pathway
Nature Communications (2019)
-
Transmembrane protein GRINA modulates aerobic glycolysis and promotes tumor progression in gastric cancer
Journal of Experimental & Clinical Cancer Research (2018)
