
Abstract
Constitutively active PI3K catalytic subunit α (PIK3CA) interfered with apoptosis induction downstream of death receptor-signaling complex formation allowing robust caspase-8 activation without triggering the execution steps of apoptosis. In mutant PIK3CA-expressing cells, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and CD95L stimulated nuclear factor kappaB (NFκB) activation, invasion, and transition to an amoeboid-like morphology. NFκB activation and adoption of amoeboid shape were inhibited by caspase-8 knockdown or FLIP-S expression, but only the cell morphology alterations required caspase-8 activity. Furthermore, we identified caspase-8-mediated, caspase-3-independent cleavage of the protein kinase rho-associated, coiled-coil containing protein kinase 1 as a novel mechanism for acquiring amoeboid shape and enhanced invasiveness in response to TRAIL and CD95L. Taken together, we provide evidence that mutated PIK3CA converts the ‘tumor surveillance’ activity of cancer cell-expressed death receptors and caspase-8 toward tumor promotion.
Similar content being viewed by others


Main
Growth factor-stimulated receptor tyrosine kinases (RTKs) typically activate the phosphoinositide 3-kinase (PI3K)/Akt pathway by recruiting inactive PI3K from the cytoplasm to phosphotyrosines in the receptor itself or in receptor-associated adaptor proteins.1, 2 Here, PI3K has improved access to its plasma membrane-located substrates and in particular converts phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 3,4,5-trisphosphate (PIP3). The latter in turn serves as a binding site for pleckstrin homology domain containing proteins, especially the serine-threonine kinase Akt/PKB and the phosphoinosite-dependent protein kinase-1 (PDK1). On activation by PDK1, Akt phosphorylates proteins involved in the regulation of proliferation, cell growth, survival, and motility, for example forkhead transcription factors, mammalian target of rapamycin, BAD, and glycogen synthase kinase-3β (GSK3β).1, 2 The major negative regulator of the PI3K pathway is the tumor suppressor phosphatase and tensin homolog (PTEN), a lipid phosphatase that dephosphorylates PIP3 and thus terminates Akt activity.1, 2
In correspondence to its crucial function in cell growth and proliferation, the PI3K/Akt/PTEN system is deregulated in a broad range of cancer entities, and mutations in its major components PI3K, PTEN, and Akt are frequently found in tumor cells. The PIP3-generating PI3Ks, which are relevant in RTK signaling, are grouped together in the IA subclass of the PI3K family and are heterodimers composed of one of the five regulatory subunits (p85α, p85β, p55α, p55γ, and p50α) and one of the three catalytic subunits (p110α, p110β, and p110γ).1, 2 In recent years, it turned out that the PI3KCA gene, which encodes the p110α catalytic subunit, is one of the most often mutated oncogenes in human malignancies. Somatic missense mutations in the PI3KCA gene have been found in liver, breast, colorectal, brain, and gastric cancer with frequencies of up to 40%.3 The mutations in the PI3KCA gene are mainly located in two hotspots encoding the helical and kinase domains of the protein and are in accordance with the concept that these alterations stimulate the lipid kinase activity of the molecule.3 In fact, it has been shown in isogenic pairs of HCT116 and DLD1 cells that harbor either the PI3KCA wild-type allele or the hotspot mutations H1047R and E545K that these genetic alterations result in improved growth factor-independent proliferation, apoptosis resistance, and tumor cell invasion.4
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor-1 (TRAILR1), TRAILR2, and CD95 are prototypic representatives of the death receptor subgroup of the TNF receptor superfamily that trigger the extrinsic pathway of apoptosis in a variety of cell types and tumor cells.5 The TRAIL death receptors and CD95 are ubiquitously expressed and their activation is mainly controlled by the strictly regulated expression of their corresponding death ligands TRAIL and CD95L. Both ligands are expressed by activated immune cells and are used to induce cell death in corresponding target cells, such as virus-infected or transformed cells.6, 7, 8 In view of the observation that late stages of some cancers, for example colorectal cancer, display constitutive expression of CD95L, which correlates with poor prognosis and enhanced metastasis,9, 10 it has been suggested that death receptor-resistant tumor cells hijack the initially anti-tumoral-acting death ligands to evade the immune system by inducing cell death in tumor-infiltrating lymphocytes.11 Additional and/or alternative pro-tumoral functions of tumor-associated CD95L expression might base on the stimulation of proinflammatory pathways.12, 13 In fact, a metastasis and invasion-promoting function has been shown in vivo for TRAIL in a model of xenotransplanted Bcl-xL-protected pancreatic ductal adenocarcinoma cells and for CD95L in a syngeneic model of intracranial glioblastoma multiforme.14, 15
Here, we show that mutated PI3K catalytic subunit α (PIK3CA) switches the quality of death receptor signaling from apoptosis induction to inflammation and invasion in colorectal HCT116 cancer cells. Particularly, we show that PIK3CA expression confers apoptosis protection downstream of death receptor-signaling complex formation and thus allows processing and activation of the complete cellular caspase-8 pool. Although caspase-8 activity is dispensable for death receptor-induced nuclear factor kappaB (NFκB) signaling, it is required to activate rho-associated, coiled-coil containing protein kinase 1 (ROCK-1), thereby inducing the remodeling of the actin cytoskeleton.
Results
Mutated PIK3CA blocks death receptor-induced apoptosis downstream of caspase-8 activation in HCT116 cells
Using gene targeting of PIK3CA in human HCT116 cancer cells, it has been shown that the exclusive expression of a single mutated PIK3CA allele harboring the activating H1047R substitution in exon 20 is sufficient to confer strong protection against TRAIL-induced apoptosis.4 In contrast, the corresponding isogenic cell line having an intact single wild-type PIK3CA allele showed almost complete cell death in response to TRAIL with an ED50-value of ∼10 ng/ml (Figure 1a). Using Fc-CD95L, a hexameric highly active variant of CD95L, we obtained similar results. HCT116 cells expressing mutated PIK3CA showed practically no apoptotic response toward Fc-CD95L, whereas the PIK3CA wild-type-expressing counterparts were readily killed at low concentrations in a caspase-dependent manner (Figure 1a and c). There were no significant differences in the cell surface expression of TRAILR1, TRAILR2, and CD95 between HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells (Figure 1d). As shown in the original description of the HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells,4 there was no or barley detectable Akt phosphorylation in the PIK3CA-wt harboring cells, whereas the HCT116-PIK3CA-mut cells displayed constitutive Akt phosphorylation (Figure 1e). In accordance with a recent study of Kleber et al.15 demonstrating CD95-induced activation of the PI3K/Akt pathway in apoptosis-resistant glioblastoma cells, we observed that stimulation with Fc-CD95L and TRAIL further increased Akt phosphorylation in HCT116-PIK3CA-mut cells (Figure 1e). Earlier reports showed that activation of the PI3K/Akt pathway can interfere with CD95L- and TRAIL-induced apoptosis by upregulation of cellular FLICE-like inhibitory proteins (cFLIP).16, 17 In case of CD95, the protective effect of PI3K/Akt signaling has additionally traced back to modulation of the lateral mobility of CD95 in the plasma membrane.18 We, therefore, next analyzed CD95-signaling complex formation and CD95 lipid raft localization in response to Fc-CD95L. In sucrose-density centrifugation, HCT116-PIK3CA-mut cells revealed a higher basal association of CD95 with the low-density lipid raft-containing fraction compared with HCT116-PIK3CA-wt cells. In both cell lines, this association was further increased in a caspase-dependent manner on stimulation with Fc-CD95L (Figure 2a). The minor PIK3CA-mut-related difference in CD95 lipid raft association was obviously of no great relevance for ligand-induced CD95-signal complex formation, as immunoprecipitation experiments demonstrated comparably efficient formation of SDS-stable high-molecular weight CD95 aggregates and also similar recruitment and processing of FADD, caspase-8, and cFLIP-L (Figure 2b). Most indicative for the unaltered caspase-8-activating capacity of CD95 and the TRAIL death receptors in the presence of PIK3CA-mut, however, was the observation that cell lysates of HCT116-PIK3CA-mut and HCT116-PIK3CA-wt cells showed no significant differences in processing of procaspase-8 to mature caspase-8 and roughly similar processing of the caspase-8 substrates FLIP-L (Figure 2b) and pro-caspase-3 (Figure 2c). Notably, there was an important functional difference in caspase-3 processing between HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells. Although there was complete processing toward the active p17 subunit of mature caspase-3 in HCT116-PIK3CA-wt cells stimulated with TRAIL or Fc-CD95L, the p19 intermediate of caspase-3 processing accumulated in HCT116-PIK3CA-mut cells (Figure 2c). Interestingly, the p19, but not the p17 subunit of caspase-3, is subject to negative regulation by IAP proteins.19 Accumulation of fully mature p17/p12 heterotetrameric caspase-3 might, therefore, be dependent on the release of pro-apoptotic mitochondrial factors, for example SMAC, that antagonize caspase inhibition by IAP proteins.20 In accordance with the idea that HCT116-PIK3CA-mut and HCT116-PIK3CA-wt cells differ in the ‘apoptotic’ activation of the mitochondria in response to death receptor activation, processing of caspase-9, caspase-2, and PARP was evident in the PIK3CA-wt-expressing cells, whereas there was only a very minor effect in this respect in the PIK3CA-mut-expressing cells (Figure 2c). Taken together, our data suggest that PIK3CA-mut blocks TRAIL- and CD95L-induced apoptosis downstream of death receptor-associated-signaling complex formation, but upstream of the release of pro-apoptotic mitochondrial factors. Indeed, a recent publication showed that expression of Bax was severely reduced in HCT116-PIK3CA-mut cells.21 We, therefore, investigated next whether the loss of Bax expression is sufficient to protect HCT116 cells from TRAIL- and CD95L-induced apoptosis. Although HCT116 cells harboring a wild type and a mutated PIK3CA allele were not as TRAIL/CD95L sensitive as HCT116-PIK3CA-wt cells, they nevertheless showed higher sensitivity than the almost completely protected HCT116-PIK3CA-mut cells (data not shown). A Bax-deficient isogenic variant derived from HCT116 cells,22 however, was almost fully rescued from death receptor-mediated apoptosis and showed a similar block of effector caspase activation downstream of death receptor-signaling-complex formation, as observed before in the HCT116-PIK3CA-mut cells (Supplementary Figure 1a and b).

PIK3CA-mut protects HCT116 cells from TRAIL- and CD95L-induced apoptosis. (a) HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells were seeded in 96-well plates and challenged the next day in triplicates with the indicated concentrations of Fc-CD95L and TRAIL. After 18 h, cell viability was determined by MTT staining. (b) Cells were incubated overnight in triplicates with the indicated mixtures of z-VAD-fmk (100 μM), Fc-CD95L (200 ng/ml), and TRAIL (50 ng/ml) and viability was again determined by MTT staining. (c) Cells were challenged with the indicated mixtures of z-VAD-fmk (100 μM), Fc-CD95L (200 ng/ml), and TRAIL (50 ng/ml) for 6 h and extracellular phosphatidylserine was detected by flow cytometry with PE-labeled annexin V. (d) Analysis of cell surface expression of TRAILR1 and TRAILR2 and CD95 by flow cytometric analysis. (e) Cells were treated with Fc-CD95L (200 ng/ml) and TRAIL (50 ng/ml) for 15 min and were analyzed with respect to Akt phosphorylation by western blotting

PIK3CA-mut rescues HCT116 cells from death receptor-induced apoptosis downstream of DISC activity. (a) HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells were stimulated with Fc-CD95L for 2 h or remained untreated. Triton × 100 lysates were prepared and subjected to cell fractionation by sucrose gradient centrifugation. The indicated fractions were analyzed by western blotting with respect to the presence of CD95, the transferrin receptor (TfR), and the lipid raft marker flotillin. Fraction 1 corresponds to the lowest sucrose density containing the detergent insoluble fraction, including microdomains and lipid rafts. The lowest, detergent soluble fraction (no. 4) contained the majority of total protein and non-lipid raft-associated receptors such as the TfR. (b) CD95-signaling complexes were induced in HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells by stimulation with 200 ng/ml Fc-CD95L for 1 and 2 h. Proteins associated with Fc-CD95L were immunoprecipitated using protein G sepharose and were analyzed together with the corresponding lysates by western blotting for the presence of CD95 and the major CD95-signaling complex components FADD, caspase-8, and cFLIP. (c) Cells were stimulated with Fc-CD95L (200 ng/ml) or TRAIL (50 ng/ml) for the indicated times and were analyzed with respect to processing of the indicated proteins by western blotting
TRAIL and CD95L elicit a proinflammatory response in PIK3CA-mut-protected HCT116 cells
TRAIL and Fc-CD95L both induced activation of the classical NFκB pathway in HCT116-PIK3CA-mut cells evident from a significant increase in inhibitor of kappaB α (IκBα) phosphorylation after 1 and 4 h post-stimulation (Figure 3a). In contrast, in HCT116-PIK3CA-wt cells, IκBα phosphorylation was poor or absent. The impaired NFκB response in TRAIL/Fc-CD95L-stimulated HCT116-PIK3CA-wt cells, however, did not reflect a fundamental difference in CD95- and TRAIL death receptor signaling between PIK3CA-wt and PIK3CA-mut-expressing cells for the following reason: caspase-8 and especially effector caspases activated in course of apoptosis shut down the classical NFκB pathway by cleavage of various NFκB-signaling intermediates.12 Accordingly, after blocking caspase activation with z-VAD-fmk in HCT116-PIK3CA-wt cells, TRAIL and Fc-CD95L induced an NFκB response as good or even better as in PIK3CA-mut-protected HCT116 cells, whereas z-VAD-fmk treatment alone had no major effect on the NFκB response (Figure 3a). Although there seemed to be quantitative differences in the extent of IκBα phosphorylation between ZVAD-protected HCT116-PIK3CA-wt and HCT116-PIK3CA-mut cells (Figure 3a), genome-wide mRNA analysis suggested that the constitutive activation of the PI3K/Akt pathway in the HCT116-PIK3CA-mut cells did not affect the upregulation of NFκB-regulated genes (Supplementary Table I). The majority of NFκB targets was similarly induced by TRAIL and Fc-CD95L and likewise upregulated in Bax-deficient HCT116 cells and parental HCT116 cells treated with z-VAD-fmk (Supplementary Table I). In accordance with the crucial function of NFκB signaling in remodeling of the extracellular matrix and metastasis, we found increased activity of TRAIL- and Fc-CD95L-stimulated HCT116-PIK3CA-mut cells in an in vitro invasion assay and enhanced uPA production (Figure 3b and c). TRAIL stimulation also enhanced invasiveness of the Bax-deficient HCT116 cells (Supplementary Figure 2).

CD95L and TRAIL elicit a pro-tumoral response in HCT116-PIK3CA-mut cells. (a) Cells were stimulated with the indicated mixtures of Fc-CD95L (200 ng/ml), TRAIL (50 ng/ml), and z-VAD-fmk (100 μM) for 1 or 4 h or remained untreated and were subsequently analyzed for the activation of the classical NFκB by western blotting using antibodies specific for phospho-IκBα and IκBα. Detection of β-actin served as loading control. (b) uPA secretion by HCT116-PIK3CA-mut cells stimulated in triplicates with Fc-CD95L (200 ng/ml) or TRAIL (50 ng/ml) for 18 h was determined by ELISA. (c) KiF-5 fibroblasts were seeded in a 24-well plate and permeabilized after 4 days with DMSO. Fibroblasts were overlayed with HCT116-PIK3CA-mut cells, which were stimulated 1 day later for 96 h with Fc-CD95L (200 ng/ml) or TRAIL (100 ng/ml). Cells were finally stained with trypan blue and photographed for documentation. The areas containing unstained cells (arrows) represent regions in which the fibroblast layer was displaced or digested by invasive tumor cells. The results shown are representative for three independent experiments
TRAIL- and CD95L-stimulated HCT116-PIK3CA-mut cells adopt an amoeba-like morphology
HCT116-PIK3CA-mut cells treated with TRAIL or Fc-CD95L rounded up and adopted a morphology resembling swollen cells undergoing necrosis (Figure 4a–d). The latter could be ruled out as the cells retained high viability (Figure 1a). Moreover, the change in morphology was reversible and disappeared within a couple of hours after removal of the initiating death ligand (data not shown) or with prolonged incubation (>48 h), (Figure 4b). Moreover, HCT116-PIK3CA-wt cells showed complete breakdown of the actin cytoskeleton in response to Fc-CD95L, whereas HCT116-PIK3CA-mut cells retained an intact actin cytoskeleton supporting the rounded cell morphology (Figure 4e). Pretreatment with z-VAD-fmk completely prevented the TRAIL- and Fc-CD95L-induced changes in cellular morphology (Figure 5a–c). This observation pointed to a possible involvement of caspase-8 in the TRAIL-/Fc-CD95L-induced alterations in cellular morphology, as this is the only caspase in HCT116-PIK3CA-mut cells that can be fully activated. Indeed, caspase-8 knockdown reduced the fraction of rounded cells in response to TRAIL and Fc-CD95L (Figure 5d). Thus, this response of HCT116-PIK3CA-mut cells was obviously dependent on caspase-8 and its enzymatic activity. Others and we have shown that CD95-induced activation of NFκB signaling requires caspase-8 independent from its enzymatic activity.23, 24 We thus also analyzed the effect of caspase-8 knockdown on Fc-CD95L- and TRAIL-induced NFκB activation in HCT116-PIK3CA-mut cells. In good accordance with our earlier studies, caspase-8 knockdown was accompanied by a strong reduction of Fc-CD95L-/TRAIL-induced upregulation of the NFκB-regulated cytokine IL8 (Figure 5e). However, as already shown in Figure 3a, inhibition of caspase-8 activity with z-VAD-fmk had no inhibitory effect. Therefore, both activation of the NFκB pathway and induction of changes in cellular morphology by Fc-CD95L and TRAIL require the presence of caspase-8, but only the latter depends additionally on the enzymatic activity of the molecule.

CD95L and TRAIL induce a change in cell morphology of non-apoptotic HCT116-PIK3CA-mut cells. (a) HCT116-PIK3CA-mut and HCT116-PIK3CA-wt cells were seeded in six-well plates (1.5 × 106 cells/well), challenged the following day with 50 ng/ml Killer-TRAIL or 200 ng/ml Fc-CD95L overnight and finally analyzed and photographed with a Zeiss Axiovert 40 inverted microscope equipped with digital camera. (b) HCT116-PIK3CA-mut cells were stimulated with TRAIL or Fc-CD95L for 12 or 48 h and cell morphology was documented by photography as in (a). (c, d) Cells were stimulated with a constant concentration of Fc-CD95L (200 ng/ml) or TRAIL (50 ng/ml) for the indicated times (c) or overnight with increasing concentrations of these ligands (d). Cells with rounded morphology were counted in three different fields. The corresponding average values are shown in the bar diagrams. (e) HCT116-PIK3CA-mut and HCT116-PIK3CA-wt cells were pretreated or not with 100 μM z-VAD-fmk and then challenged for 3 h with Fc-CD95L (200 ng/ml). After fixation, actin was stained with AlexaFluor488-labeled phalloidin

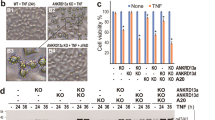
Caspase-8 is required for CD95L- and TRAIL-induced alterations in cell morphology of non-apoptotic HCT116-PIK3CA-mut cells. (a–c) HCT116-PIK3CA-mut cells were treated or not with 100 μM of z-VAD-fmk 1 h prior stimulation with death ligands (Fc-CD95L: 200 ng/ml; TRAIL: 50 ng/ml). Cells were photographed the next day with a Zeiss Axiovert 40 inverted microscope equipped with digital camera (a) or stained with AlexaFluor488-labeled phalloidin after fixation and analyzed by confocal fluorescence microscopy (b) Again the percentage of cells with rounded morphology was calculated from three independent fields and the corresponding average values are shown in the bar diagram (c). (d) HCT116-PIK3CA-mut cells were transfected with the indicated siRNA oligonucleotides. After 48 h, cells were analyzed by western blotting for the presence of caspase-8. Cells with rounded morphology were scored again by microscopic inspection. (e) Caspase-8 expression was knocked down in HCT116-PIK3CA-mut cells as before. Cells were analyzed with respect to Fc-CD95L-induced IL8 production by ELISA
TRAIL- and CD95L-induced changes in cellular morphology depend on caspase-8-mediated activation of ROCK-1
The type of TRAIL- and Fc-CD95L-induced morphological changes in HCT116-PIK3CA-mut cells points to an involvement of the actin cytoskeleton. ROCK-1 has a central function in the regulation of the actin cytoskeleton and is a prominent caspase-3 substrate that is cleaved and activated during apoptosis.25, 26 Activation of ROCK-1 is crucial for apoptosis-accompanied membrane blebbing, a classical hallmark of the apoptotic execution phase.25, 26 As the shown caspase-3 cleavage site of ROCK-1 (DETD1113) matches also reasonable well with prototypical caspase-8 recognition sites (Figure 6a), we evaluated next the possible involvement of ROCK-1 in the cell death-independent changes in cell morphology induced by TRAIL and Fc-CD95L. ROCK-1 was cleaved with comparable efficacy in HCT116-PIK3CA-wt cells undergoing apoptosis and apoptosis-resistant HCT116-PIK3CA-mut cells (Figure 6b), suggesting that ROCK-1 cleavage is independent from active effector caspases and apoptosis. Moreover, a selective caspase-3 inhibitor (DQMD-fmk) partially rescued HCT116-PIK3CA-wt cells from TRAIL- and Fc-CD95L-induced apoptosis, but showed no major effect on ROCK-1 cleavage and reorganization of cellular shape in HCT116-PIK3CA-mut cells (Figure 6c–e). Notably, when the sensitive HCT116-PIK3CA-wt cells were rescued from TRAIL/FasL-induced apoptosis with DQMD-fmk, these cells also adopt an amoeboid shape (data not shown). In further accordance with a function of activating ROCK-1 cleavage in TRAIL-/Fc-CD95L-induced amoeboid morphology, the fraction of HCT116-PIK3CA-mut cells with rounded shape was significantly reduced on pretreatment with the ROCK-1 inhibitor Y27632 or ROCK-1 knockdown (Figure 7a and b). TRAIL- and Fc-CD95L-induced NFκB signaling, however, was independent from ROCK-1 (data not shown). Notably, pharmacological ROCK-1 inhibition almost completely inhibited the TRAIL- and Fc-CD95L-induced invasive activity of HCT116-PIK3CA-mut cells (Figure 7c). Pretreatment with Y27632 neither exerted a significant effect on apoptosis sensitivity of HCT116-PIK3CA-wt cells (Supplementary Figure 3a and b) nor it sensitized HCT116-PIK3CA-mut cells for death receptor-induced apoptosis (Supplementary Figure 3b). Thus, ROCK-1 is involved in a subset of non-apoptotic death receptor-induced cellular effects, but is dispensable for apoptosis induction.

TRAIL- and CD95L-induced ROCK-1 cleavage under non-apoptotic conditions contributes to changes in cell morphology. (a) Comparison of the reported caspase cleavage site in ROCK-1 with cleavage sites of well-established substrates of caspase-3 and -8. The scoring of the cleavage sites was performed with the GraBCas bioinformatics tool.37 (b) HCT116-PIK3CA-mut and HCT116-PIK3CA-wt cells were challenged with Fc-CD95L (200 ng/ml) or TRAIL (50 ng/ml) and subjected to western blot analysis for the detection of processing of ROCK-1. ‘n.s.’ indicates non-specific background from ECL detection. (c) HCT116-PIK3CA-wt cells were stimulated overnight in triplicates with 50 ng/ml Killer-TRAIL or 200 ng/ml Fc-CD95L in the presence or absence of the caspase-3 inhibitor DQMD-fmk (20 μM). Cell viability was finally determined by MTT staining (left panel). (d) HCT116-PIK3CA-mut cells were stimulated with TRAIL and Fc-CD95L in the presence and absence of DQMD-fmk (20 μM) and analyzed by western blotting with respect to ROCK-1 cleavage and caspase-3 processing. (e) HCT116-PIK3CA-mut cells were pretreated with 20 μM of the caspase-3-specific inhibitor DQMD-fmk and were subsequently challenged with 50 ng/ml of Killer-TRAIL or 200 ng/ml Fc-CD95L. The fraction of cells with rounded, amoeboid shape was determined by microscopic inspection

ROCK-1 activity is required for TRAIL- and CD95L-induced cellular remodeling and invasion. (a) HCT116-PIK3CA-mut cells were pretreated with 20 μM of the ROCK-1 inhibitor Y27632 and were subsequently challenged with 50 ng/ml of Killer-TRAIL or 200 ng/ml Fc-CD95L. The fraction of cells with rounded, amoeboid shape was determined by microscopic inspection. (b) ROCK-1 expression was knocked down in HCT116-PIK3CA-mut cells. Cells were then analyzed with respect to the efficacy of inhibition of ROCK-1 expression by western blotting or were subjected to stimulation experiments with Fc-CD95L and Killer-TRAIL. The fraction of cells with rounded amoeboid shape was again determined by microscopic inspection. (c) A KiF-5 fibroblasts layer were permeabilized with DMSO and overlayed with HCT116-PIK3CA-mut cells, which were stimulated 1 day later for 96 h with Fc-CD95L (200 ng/ml) or TRAIL (100 ng/ml) with or without concomitant treatment with Y27632 (20 μM). Cells were stained with trypan blue and photographed for documentation. The areas containing unstained cells (arrows) represent regions in which the fibroblast layer was displaced or digested by invasive HCT116-PIK3CA-mut cells
cFLIP-S inhibits induction of amoeboid cellular morphology by TRAIL and CD95L
In agreement with our aforementioned observation that caspase-8 activity is required for TRAIL-/Fc-CD95L-induced alterations in cellular morphology, we further found that ectopic expression of cFLIP-S not only prevented death receptor-associated caspase-8 maturation, but also death receptor-induced remodeling of the cytoskeleton in non-apoptotic HCT116-PIK3CA-mut cells (Figure 8a and b). FLIP proteins also interfere with NFκB activation by TRAIL and Fc-CD95L.23, 24, 27, 28 Consistent with this, we noticed that cFLIP-S expression abrogated TRAIL- and Fc-CD95L-induced upregulation of the NFκB-regulated target IL8 in HCT116-PIK3CA-mut cells (Figure 8c). As cFLIP proteins can contribute to tumor cell escape from apoptosis induction by death receptors, these proteins are commonly regarded as tumor-promoting factors. In contrast, our cFLIP-related data now suggest that cFLIP proteins can also exert anti-tumoral effects in death receptor-resistant tumor cells.

cFLIP-S blocks non-apoptotic signaling by CD95 and the TRAIL death receptors. (a) HCT116-PIK3CA-mut cells transfected with an cFLIP-S encoding expression vector and the corresponding control plasmid were analyzed with respect to cFLIP-S expression and TRAIL- and Fc-CD95L-induced caspase-8 processing by western blotting. (b) Cells were analyzed with respect to TRAIL- and Fc-CD95L-induced rounding of cells as described in the earlier figures. (c) cFLIP-S transfectants and their corresponding control cells were stimulated with Killer-TRAIL (50 ng/ml) and Fc-CD95L (200 ng/ml). Cells were stimulated for 6 h and IL8 production was determined by ELISA
Discussion
In this study, we analyzed the cooperation of the prominent oncogenic PI3K/Akt-signaling pathway with CD95 and the TRAIL death receptors. Analysis of isogenic HCT116 cell lines expressing wild-type or mutant PIK3CA revealed enhanced growth factor-independent cell proliferation and resistance against apoptosis induction by TRAIL and growth factor deprivation in the mutant PIK3CA cells.4, 21 In accordance with the study of Guo et al.,21 which traced back apoptosis resistance of growth factor-deprived HCT116-PIK3CA-mut cells to downregulation of Bax expression, we found no major differences in the recruitment and activation of caspase-8 by CD95 and TRAIL death receptor between wild-type and mutant PIK3CA-expressing isogenic HCT116 cells (Figures 1 and 2). Thus, resistance against death receptor-induced apoptosis in the HCT116-PIK3CA-mut model is largely dependent on mechanisms acting downstream of death receptor-associated caspase-8 maturation. In other cellular systems, PI3K/Akt pathway-dependent death receptor resistance has been assigned to changes in receptor mobility in the plasma membrane or upregulation of the caspase-8 inhibitory cFLIP proteins.16, 17, 18 Our data thus suggest that the PTEN-PI3K-Akt system can confer protection against death receptor apoptosis by independently acting mechanisms in a cell type-specific manner. The plasticity in anti-apoptotic PI3K-Akt signaling might become especially important in view of the fact that distinct apoptosis inhibitory mechanisms differentially affect non-apoptotic signaling by CD95 and the TRAIL death receptors. Indeed, we observed that expression of cFLIP-S, which is a prominent and strong inhibitor of CD95- and TRAIL-induced apoptosis, also blocks TRAIL- and Fc-CD95L-stimulated NFκB activation in apoptosis-resistant cells expressing mutated PIK3CA (Figure 8). Thus, dependent on how the PTEN-PI3K-Akt system mediates death receptor resistance, it not only supports tumor cell survival by preventing TRAIL- and Fc-CD95L-induced apoptosis, but in addition enables cancer cells to hijack these molecules to establish a tumor-promoting microenvironment.
An involvement of CD95 in driving invasion and metastasis of cancer cells has recently been shown for glioblastoma cells.15 In this study, activation of CD95 resulted in recruitment and activation of Src family kinases and the p85 subunit of PI3K accompanied by a subsequent increase in invasiveness because of stimulation of the Akt-GSK3β-β-catenin axis and upregulation of matrix metalloproteinases.15 Remarkably, CD95-induced PI3K activation and cell migration in glioblastoma cells occur in a manner that is neither blocked by the general caspase inhibitor z-VAD-fmk nor accompanied by increased recruitment of FADD and caspase-8 to CD95.
There is evidence that caspase-8 also has a function in death receptor-independent modes of cell migration and cell motility. So, in NB7 neuroblastoma cells with hyperactive Src or stimulated with EGF, caspase-8 becomes phosphorylated on Y380 enabling the protein to bind the p85 subunit of PI3K and to activate the PI3K effector Rac, which is of pivotal relevance for cell migration.29, 30 In this model of cell migration, the enzymatic activity of caspase-8 is again dispensable.29, 30 In the same cellular model, a non-catalytic function of caspase-8 in Src-dependent cell adhesion and activation of extracellular-regulated kinase 1/2 has also been shown, but the relation of these events to p85/Rac-mediated cell migration is still unclear.31 In addition, caspase-8 has also been found to be recruited to the focal adhesion complex in NB7 cells in which it enhances cell migration by stimulating talin cleavage by calpains in a caspase-activity/apoptosis-independent manner.32, 33 As Src and PI3K are part of the focal adhesion complex, it is tempting to speculate that Y380 phosphorylation of caspase-8 and activation of PI3K/Rac reflect a function of caspase-8 in the focal adhesion complex. Taken together, to date, there is no evidence that the migration-promoting functions of CD95 and/or caspase-8 require caspase-8 activity.
Now, we observed that TRAIL and Fc-CD95L induce transition to an amoeboid-like cell shape that has been associated in recent years with a specialized type of cell migration that differs from the classical ‘mesenchymal’ type of cell migration regarded above. Although the mesenchymal type of cell migration requires pericellular proteolysis and Rac-mediated reorganization of the actin cytoskeleton, the amoeboid type of cell invasion is independent from pericellular proteolysis and has crucial requirement for ROCK-1 signaling.34, 35 Noteworthy, the TRAIL-/Fc-CD95L-induced transition to an amoeboid cell morphology described in our study is not only dependent on caspase-8 and its enzymatic activity, but also involves ROCK activity (Figures 5a–c, 7a, and b). Moreover, it has been earlier shown that in course of apoptosis, caspase-3-mediated cleavage of ROCK-1 releases an active fragment that induces apoptotic membrane blebbing.25, 26 In good accordance with the idea that CD95 and the TRAIL death receptors trigger amoeboid transition by caspase-8-mediated, caspase-3-independent cleavage of ROCK-1 in non-apoptotic cells by a related mechanism, we showed that ROCK-1 cleavage and amoeboid transition are not significantly affected by caspase-3 inhibition and/or expression of PIK3CA-mut (Figure 6b, d, and e). Notably, the inhibitory effect of caspase-8 knockdown on the death receptor-induced morphological change in cellular shape was more pronounced compared with the effect of ROCK-1 knockdown (Figures 5d and 7b). This might be due to the existence of additional caspase-8 substrates affecting the cellular shape. In fact, caspase-8 also cleaves plectin, which links the actin cytoskeleton to microtubules and intermediate filaments.36
Fc-CD95L also induced an amoeboid morphology in apoptosis-resistant HCT116-Bax−/− cells. However, in these cells, the effect was less pronounced than in the HCT116-PIK3CA-mut cells (Supplementary Figure 4; Figure 4). Parental HCT116 cells, which harbor a PIK3CA-wt and a PIK3CA-mut allele, are almost as sensitive for apoptosis induction as HCT116-PIK3CA-wt cells (Supplementary Figure 1a; Figure 1). Thus, in a situation of mixed PIK3CA-mut and PIK3CA-wt alleles, the latter (wt) seems to dominate the cellular effects. The finding of a reduced Fc-CD95L-induced adoption of amoeboid shape in the HCT116-Bax−/− cells compared with the equally resistant HCT116-PIK3CA-mut cells might, therefore, mean that not only caspase-8-mediated ROCK-1 cleavage is involved in triggering the adaptation of amoeboid shape, but also other PI3K/Akt pathway-dependent mechanisms. In fact, IGF-1, which activates the PI3K/Akt pathway, also increased the number of HCT116-Bax−/− cells with amoeboid shape ∼twofold (Supplementary Figure 4).
We also analyzed whether HCT116-PIK3CA-mut cells undergo transition of a spindle shaped to amoeboid migration in a 3D collagen lattice in response to TRAIL or Fc-CD95L. However, the HCT116-PIK3CA-mut cells proved to be not well suited to this assay. First, the HCT116-PIK3CA-mut cells needed around 2 days to readopt their flat shape in the 3D collagen lattice, and second, showed poor migration and no significant alterations in cellular morphology irrespective of TRAIL treatment (data not shown). Therefore, further investigations are under way to answer the question whether death ligand-induced adoption of an amoboid cellular shape is sufficient for the amoboid-like type of migration.
Materials and Methods
Cell lines, reagents, and antibodies
HCT116-PIK3CA-wt, HCT116-PIK3CA-mut, and HCT116-Bax-/- were a kind gift of B Vogelstein (Johns Hopkins University, Baltimore, USA) and maintained in RPMI1640 medium (PAA, Pasching, Austria) containing 10% heat inactivated fetal calf serum (PAA). Fc-CD95L was produced in HEK293 cells and was purified by anti-Flag mAb affinity chromatography taking advantage of an internal Flag epitope. Protein G agarose beads were purchased from Sigma (Deisenhofen, Germany), z-VAD-fmk from Bachem (Heidelberg, Germany), and Killer-TRAIL from Alexis (Lörrach, Germany). Antibodies specific for caspase-3, caspase-9, JNK, phospho-JNK, p44/42, phospho-p44/42, p38, phospho-p38, and phospho-IκBα were purchased from Cell Signaling (Beverly, MA, USA). IκBα-, CD95-, and FADD-specific antibodies were obtained from Santa Cruz (Santa Cruz, CA, USA); anti-RIP, anti-caspase-2, anti-transferrin receptor, anti-flotillin, and anti-PARP1 were from BD Biosciences Pharmingen (Heidelberg, Germany); anti-β-actin was obtained from Sigma; and anti-FLIP from Alexis.
Cell death assay
Cells (20 × 103 per well) were seeded in 96-well plates. The next day, cells were challenged with indicated concentrations of Fc-CD95L or Killer-TRAIL in triplicates. Cell viability was determined after 18 h using MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) staining.
Western blotting
For detection of phosphorylated proteins, cells were harvested in medium, spun down, and directly dissolved in 4 × Laemmli sample buffer (8% SDS, 0.1M dithiothreitol, 40% glycerol, 0.2 M Tris, pH 8.0) supplemented with phosphatase inhibitor cocktails-I and -II (Sigma). Lysates were sonicated and subsequently boiled for 5 min at 96°C. For western blot analyses, proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and non-specific binding sites were blocked by incubation in Tris-buffered saline containing 0.1% Tween 20 and 5% dry milk. Membranes were incubated with primary antibodies of the indicated specificity, followed by horseradish peroxidase-conjugated secondary antibodies (Dako, Hamburg, Germany). Protein bands were visualized using ECL western blotting detection reagents (Amersham Biosciences Europe, Freiburg, Germany).
For preparation of triton × 100 lysates, cells were washed, resuspended in lysis buffer containing 30 mM Tris–HCl, 1% triton × 100, 10% glycerol, 120 mM NaCl, pH 7.5, supplemented with complete protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany), and incubated 20 min on ice. Lysates were cleared by centrifugation (20 min, 14.000 × g), Laemmli sample buffer was added, and samples were boiled for 5 min at 96°C.
Isolation of detergent insoluble membrane fractions
A total of 3 × 107 cells per group were harvested, suspended in 200 μl serum-free RPMI 1640 medium and lysed in 200 μl ice-cold triton × 100 lysis buffer (0.8% triton × 100 in TNE (25 mM Tris, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM Pefabloc, 5 mM C2H4INO, 1 mM Na3VO4, 1 mM NaF)). Lysates were loaded on discontinuous sucrose gradients and after centrifugation at 50 000 r.p.m. in a Beckman SW60 rotor for 22 h at 4°C, four fractions were collected from each sample and analyzed by western blotting. Detection of the established lipid raft marker flotillin was used to identify the lipid raft-containing fraction.
Flow cytometry and annexin V staining
Cells were incubated for 30 min on ice with PE-conjugated TRAILR1-/TRAILR2-specific antibody, FITC-conjugated anti-CD95 antibody (R&D Systems) or an appropriate isotype control (R&D Systems). Annexin V was purchased from Immunotools (Friesoythe, Germany) and used according to manufacturers’ instructions. Analyses were performed using FACSCalibur (BD Biosciences, Heidelberg, Germany) following standard procedures.
Coimmunoprecipitation
Immunoprecipitation of CD95 was performed with Fc-CD95L using one confluent 175 cm2 flask of cells per condition. Briefly, cells were stimulated with 200 ng/ml Fc-CD95L in 5 ml medium for the indicated times at 37°C, washed in ice-cold PBS, transferred in 1.5 ml lysis buffer (30 mM Tris–HCl, pH 7.5, 1% triton × 100, 10% glycerol, 120 mM NaCl) supplemented with complete protease inhibitor cocktail (Roche Diagnostics GmbH), and incubated for 20 min on ice. Lysates were cleared by centrifugation (2 × 20 min, 14 000 g) and CD95 complexes were precipitated with protein G agarose (40 μl of a 50% slurry) at 4°C overnight. Lysates from unstimulated cells supplemented with 50 ng of Fc-CD95L before adding protein G beads served as negative control. After washing in lysis buffer, agarose-bound proteins were eluted by incubation at 75°C for 10 min in 4 × Laemmli sample buffer.
Determination of IL8 and uPA production
For determination of IL8 secretion, cells (20 × 103 per well) were seeded in triplicates in 96-well tissue culture plates and cultured overnight. Thereafter, medium was changed and the indicated concentrations of Fc-CD95L or Killer-TRAIL or medium were added for 4 h in the presence or absence of 100 μM z-VAD-fmk. IL8 was quantified in the collected supernatants using an enzyme-linked immunosorbent assay (BD Biosciences Pharmingen). For quantification of uPA in cell culture, supernatants cells were seeded in 96-well tissue culture plates and stimulated next day with Fc-CD95L and Killer-TRAIL for 18 h. The amounts of uPA in cell culture supernatants were determined by uPA ELISA (American Diagnostica, Greenwich, CT, USA). Concentration of uPA was normalized to the cell number determined in parallel.
siRNA experiments
Knockdown of caspase-8 or ROCK-1 was performed using specific siRNA (Qiagen, Hilden, Germany). Briefly, cells were seeded in six-well plates (2.5 × 105 cells per well) and after incubation over night, 100 pmol of specific or control siRNA were transfected using Lipofectamine 2000 transfection system (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Two days post-transfection, knockdown efficacy was determined by western blot analysis and subsequent treatment started. Incubation with Fc-CD95L (200 ng/ml) or Killer-TRAIL (50 ng/ml) lasted over night for microscopy analysis (see below) or 4 h for measuring IL8 induction.
Microscopy
Light microscopy analyses were performed using a Zeiss Axiovert 40 inverted microscope (Zeiss, Oberkochen, Germany), connected to a Canon Powershot A620 digital camera (Canon, Krefeld, Germany). For morphologic analyses of HCT116-PIK3CA-mut cells, cells were seeded in six-well plates (1.5 × 106 cells per well). The next day, cells underwent treatment with Killer-TRAIL or Fc-CD95L, lasting over night. In case of preceding siRNA transfection, treatment started 48 h thereafter, lasting again over night. Pictures were taken using ‘Psremote’ software (Breeze Systems, Bagshot, UK). For analysis, cells displaying round morphology were counted in three randomly selected fields ( × 400 magnification). Statistical and graphic analyses were performed using the Software Graph Pad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). For visualization of F-actin, cells were grown on glass coverslips for and treated as indicated. After fixation with 2% formaldehyde (freshly prepared from paraformaldehyde) in PBS (10 min, RT), cells were immersed in PBS containing 0.1% triton × 100 for 5 min at room temperature. After incubation with 10% normal goat serum and 1% bovine serum albumin (30 min, RT), F-actin was stained with ALEXA488-labeled phalloidin (Mobitec, Goettingen, Germany) for 60 min at room temperature. Coverslips were rinsed with PBS (5 min, three times each) and mounted on glass slides with 60% glycerol in PBS containing 1.5% n-propyl gallate (Serva, Heidelberg, Germany) as anti-fading compound. A confocal laser scanning microscope (LSM510, Carl Zeiss Microimaging, Goettingen, Germany) was used for analysis and imaging.
Invasion assay
To evaluate the invasive potential of HCT116-PIK3CA-mut cells, 2.5 × 105 KiF-5 fibroblasts were seeded per well of a 24-well plate, cultivated for 4 days, washed with PBS, and permeabilized by adding 500 μl DMSO per well for 1 h at room temperature. After washing with PBS, the fibroblasts were overlayed with HCT116-PIK3CA-mut cells (2 × 104 cells/well in culture medium). After 24 h, medium was replaced by medium alone or medium supplemented with Killer-TRAIL (50 ng/ml) or Fc-CD95L (200 ng/ml). After additional 2 days, cells were rinsed with PBS and stained by incubation with 0.2% trypan blue for 15 min. After removing the staining solution and washing twice with PBS, cells were finally photographed. The living HCT116-PIK3CA-mut cells could be distinguished from the KiF-5 fibroblasts layer as trypan blue preferentially stain the latter cells, which are dead and permeabilized. Areas covered by unstained cells thus represent regions in which fibroblasts were displaced or digested by invasive HCT116-PIK3CA-mut cells.
Abbreviations
- IκB:
-
inhibitor of kappaB
- NFκB:
-
nuclear factor kappaB
- PI3K:
-
phosphoinositide 3-kinase
- PIK3CA :
-
PI3K catalytic subunit α
- ROCK:
-
Rho-associated, coiled-coil containing protein kinase 1
- TRAIL:
-
TNF-related apoptosis-inducing ligand
References
Yuan TL, Cantley LC . PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27: 5497–5510.
Zhao L, Vogt PK . Class I PI3K in oncogenic cellular transformation. Oncogene 2008; 27: 5486–5496.
Karakas B, Bachman KE, Park BH . Mutation of the PIK3CA oncogene in human cancers. Br J Cancer 2006; 94: 455–459.
Samuels Y, Diaz Jr LA, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005; 7: 561–573.
Wajant H . Death receptors. Essays Biochem 2003; 39: 53–71.6.
Wajant H . CD95L/FasL and TRAIL in tumour surveillance and cancer therapy. Cancer Treat Res 2006; 130: 141–165.
Keckler MS . Dodging the CTL response: viral evasion of Fas and granzyme induced apoptosis. Front Biosci 2007; 12: 725–732.
Strater J, Moller P . TRAIL and viral infection. Vitam Horm 2004; 67: 257–274.
Okada K, Komuta K, Hashimoto S, Matsuzaki S, Kanematsu T, Koji T . Frequency of apoptosis of tumor-infiltrating lymphocytes induced by fas counterattack in human colorectal carcinoma and its correlation with prognosis. Clin Cancer Res 2000; 6: 3560–3564.
Yokomizo H, Yoshimatsu K, Ishibashi K, Hashimoto M, Yosida K, Kato H et al. Fas ligand expression is a risk factor for liver metastasis in colorectal cancer with venous invasion. Anticancer Res 2003; 23: 5221–5224.
Igney FH, Krammer PH . Tumor counterattack: fact or fiction? Cancer Immunol Immunother 2005; 54: 1127–1136.
Wajant H, Pfizenmaier K, Scheurich P . Non-apoptotic Fas signaling. Cytokine Growth Factor Rev 2003; 14: 53–66.
Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK et al. The CD95 receptor: apoptosis revisited. Cell 2007; 129: 447–450.
Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D et al. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006; 25: 7434–7439.
Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008; 13: 235–248.
Suhara T, Mano T, Oliveira BE, Walsh K . Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP). Circ Res 2001; 89: 13–19.
Panka DJ, Mano T, Suhara T, Walsh K, Mier JW . Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J Biol Chem 2001; 276: 6893–6896.
Varadhachary AS, Edidin M, Hanlon AM, Peter ME, Krammer PH, Salgame P . Phosphatidylinositol 3′-kinase blocks CD95 aggregation and caspase-8 cleavage at the death-inducing signaling complex by modulating lateral diffusion of CD95. J Immunol 2001; 166: 6564–6569.
Choi YE, Butterworth M, Malladi S, Duckett CS, Cohen GM, Bratton SB . The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J Biol Chem 2009; 284: 12772–12782.
Vaux DL, Silke J . Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun 2003; 304: 499–504.
Guo XN, Rajput A, Rose R, Hauser J, Beko A, Kuropatwinski K et al. Mutant PIK3CA-bearing colon cancer cells display increased metastasis in an orthotopic model. Cancer Res 2007; 67: 5851–5858.
Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B . Role of BAX in the apoptotic response to anticancer agents. Science 2000; 290: 989–992.
Kreuz S, Siegmund D, Rumpf JJ, Samel D, Leverkus M, Janssen O et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol 2004; 166: 369–380.
Imamura R, Konaka K, Matsumoto N, Hasegawa M, Fukui M, Mukaida N et al. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem 2004; 279: 46415–46423.
Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF . Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 2001; 3: 339–345.
Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J . Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol 2001; 3: 346–352.
Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G et al. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J Biol Chem 2000; 275: 24357–24366.
Wachter T, Sprick M, Hausmann D, Kerstan A, McPherson K, Stassi G et al. cFLIPL inhibits tumor necrosis factor-related apoptosis-inducing ligand-mediated NF-kappaB activation at the death-inducing signaling complex in human keratinocytes. J Biol Chem 2004; 279: 52824–52834.
Senft J, Helfer B, Frisch SM . Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res 2007; 67: 11505–11509.
Torres VA, Mielgo A, Barila D, Anderson DH, Stupack D . Caspase 8 promotes peripheral localization and activation of Rab5. J Biol Chem 2008; 283: 36280–36289.
Finlay D, Vuori K . Novel noncatalytic role for caspase-8 in promoting SRC-mediated adhesion and Erk signaling in neuroblastoma cells. Cancer Res 2007; 67: 11704–11711.
Helfer B, Boswell BC, Finlay D, Cipres A, Vuori K, Bong Kang T et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res 2006; 66: 4273–4278.
Barbero S, Mielgo A, Torres V, Teitz T, Shields DJ, Mikolon D et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res 2009; 69: 3755–3763.
Sahai E, Marshall CJ . Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 2003; 5: 711–719.
Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003; 160: 267–277.
Stegh AH, Herrmann H, Lampel S, Weisenberger D, Andra K, Seper M et al. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol Cell Biol 2000; 20: 5665–5679.
Backes C, Kuentzer J, Lenhof HP, Comtesse N, Meese E . GraBCas: a bioinformatics tool for score-based prediction of Caspase- and Granzyme B-cleavage sites in protein sequences. Nucleic Acids Res 2005; 33: W208–W213.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG Wa 1025/18-1, SFB487 project B7 and SFB415 project A3) and Deutsche Krebshilfe (project 107034-226). ME was supported by an MD/PhD fellowship of the IZKF Würzburg. We thank Prof. Peter Friedl (Department of Cell Biology (283), Radboud University Nijmegen, The Netherlands) and Margit Ott (Department of Dermatology, University Hospital Würzburg, Germany) most sincerely for analyzing morphology of HCT116-PIK3CA-mut cells in 3D collagen lattice. We are also indebted to Bert Vogelstein (The John Hopkins University School of Medicine, USA) for kindly providing HCT116-PIK3CA-mut, HCT116-PIK3CA-wt, HCT116-Bax-/-, and parental HCT116 cells.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Additional information
Edited by J Tschopp
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Ehrenschwender, M., Siegmund, D., Wicovsky, A. et al. Mutant PIK3CA licenses TRAIL and CD95L to induce non-apoptotic caspase-8-mediated ROCK activation. Cell Death Differ 17, 1435–1447 (2010). https://doi.org/10.1038/cdd.2010.36
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.36
Keywords
This article is cited by
-
Harnessing TRAIL-induced cell death for cancer therapy: a long walk with thrilling discoveries
Cell Death & Differentiation (2023)
-
A fate worse than death: apoptosis as an oncogenic process
Nature Reviews Cancer (2016)
-
Targeting caspase-6 and caspase-8 to promote neuronal survival following ischemic stroke
Cell Death & Disease (2015)
-
Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1
Cell Death & Differentiation (2014)
-
XIAP-targeting drugs re-sensitize PIK3CA-mutated colorectal cancer cells for death receptor-induced apoptosis
Cell Death & Disease (2014)
