
Abstract
Background:
No blood biomarkers to detect early oral cavity squamous cell carcinoma (OSCC) without clinical signs exist – diagnosis is solely based on histology of a visible tumour. Most OSCC patients are diagnosed at advanced stage, which leads to significant morbidity and poor survival. Our aim was to find the serum screening or detection biomarkers in OSCC.
Methods:
Serum samples from patients with OSCC treated at the Department of Otorhinolaryngology – Head and Neck Surgery, Helsinki University Hospital (Finland) were collected. Age- and gender-matched healthy individuals served as controls. Quantitative label-free proteomics in high definition MSE mode(HDMSE) was performed on 13 patients and 12 healthy samples. Various statistical analyses were performed on quantitative proteomics data to obtain the most influential proteins, which classify the patients vs healthy samples.
Results:
In quantitative proteomic analysis (HDMSE), 388 proteins were quantified in our pilot study. A complete separation between cases and controls was seen in supervised and unsupervised classification techniques such as orthogonal projections on latent structure-discriminant analysis (OPLS-DA) and self-organising maps. Using OPLS-DA S-plot, we identified a set of eight proteins that completely separated OSCC patients from healthy individuals.
Conclusions:
Although the tumour stages varied from I to IVa, these potential biomarkers were able to identify all OSCCs demonstrating their sensitivity to detect tumours of all stages. We are the first to suggest a set of serum biomarkers in our pilot study to be evaluated further as a diagnostic panel to detect preclinical OSCC in risk patients.
Similar content being viewed by others


Main
Head and neck squamous cell carcinomas (HNSCCs) include a heterogeneous group of neoplasms with different tumour biologies, response to therapies and prognosis (Razzouk, 2014). Smoking and alcohol consumption are the most common aetiological risk factors (Wynder et al, 1977; Smith, 1979; Decker and Goldstein, 1982). Despite recent improvements in treatment and utilisation of multimodal treatment (surgery, radiotherapy, chemotherapy, alone or in combination), the outcome of HNSCC patients’ remains poor, and 40 to 60% of the patients with locally advanced HNSCC experience recurrent disease (Machiels et al, 2014).
The incidence of oral cavity squamous cell carcinoma (OSCC) has increased during the last decade making it the most common type of head and neck cancer (HNC) of which the most common is the laryngeal cancer (Bagan and Scully, 2008; Chaturvedi et al, 2013; Braakhuis et al, 2014; van Dijk et al, 2016). The increase has been most significant among tongue cancers (van Dijk et al, 2016). At the moment, the diagnostic tools available for tumours in the oral cavity are clinical examination together with tissue biopsies. The big advantages of serum biomarkers are their minimal invasiveness and easiness of testing, when comparing to tissue biopsies. Among heavy smokers and drinkers, these biomarkers might even be tested annually, to screen for OSCC or, for example, if patient is having unspecific symptoms in the oral cavity, and no visible lesions are available to take a biopsy. High-throughput plasma/serum proteomics is a method enabling the discovery of target biomarkers with high sensitivity to separate different biological states.
Aiming to find novel biomarkers, we have used high definition MSE (HDMSE) methodology to analyse the serum samples of OSCC patients and their age-matched healthy controls. The target was to separate the two groups and to identify protein attributes enabling this separation. To find the ability of proteomic analysis to classify the samples into two separate groups, three parallel methods were used to separate OSCC samples from healthy control. Two of these methods were unsupervised, including hierarchical clustering and self-organising maps (SOMs) while the third was supervised separation method called orthogonal projections to latent structure-discriminant analysis (OPLS-DA). In addition to the protein abundancies, we wanted to study the overrepresented biological pathways in the two groups.
Materials and methods
Serum samples
Serum samples were collected from patients with OSCC treated at the Department of Otorhinolaryngology – Head and Neck Surgery, Helsinki University Hospital (Helsinki, Finland). An approval for the study was received from the Ethics Committee of Medical Sciences (Dnro: 64/13/03/02/2014) and a written informed consent was obtained from all subjects at the time of serum sample collection. Serum samples were stored at −70 °C until tested at the same time. All experiments were performed in accordance with relevant guidelines and regulations. In all, 13 patients and 12 healthy control samples were used in the study.
Reagents
For serum preprocessing Pierce SwellGel Blue Albumin Removal Discs, Pierce Centrifuge columns as well as Pierce C18 Spin Columns were purchased from Thermo Scientific (Rockform, IL, USA). The solvents and high-purity HPLC reagents were purchased from Waters (Milford, MA, USA). All other reagents were from Sigma-Aldrich (St Louis, MO, USA).
Serum treatment and protein digestion
The workflow as well as serum treatment and protein digestion are described in detail previously (Kuusela et al, 2017). Briefly, after thawing of samples top 12 proteins of serum were depleted by Pierce Top 12 protein depletion columns according to the manufacturer’s instructions. Top 12 protein-depleted serum was assayed by bicinchoninic acid assay kit (Pierce, Thermo Scientific, Rockform, IL, USA) for the total protein concentration. Top 12 protein-depleted serum corresponding to 350 μg of protein was dried in speed vacuum system (Savant, Thermofisher). After dissolving the dried pellets in 6 M urea and 100 mM Tris-HCl (pH 7.4), reduction and alkylation were performed by incubating samples first in 10 mM of dithiothreitol (DTT) for 60 min at room temperature (RT) followed by incubation in 30 mM iodoacetamide for 60 min in the dark at RT. Finally, after consumption of excess of iodoacetamide with 30 mM DTT (60 min at RT) samples were diluted 1:10 with high-purity Milli-Q water (Millipore, Billerica, MA, USA) and digested with trypsin (1:50 trypsin to protein) for 18 h at +37 °C. Samples were cleaned by Pierce C18 columns according to the manufacture’s protocol. For liquid chromatography–mass spectrometry (LC–MS) analysis 30 μg of peptides were first dried in speed vacuum drier, dissolved in 0.1% formic acid containing 12.5 fmol Hi3 peptide mixture (Waters) per μl and finally stored at −20 °C until analysed.
LC–MS and quantification ultra performance liquid chromatography–MS
Four microlitre samples, equivalent to ∼1.4 μg total protein, were injected to nano Acquity UPLC system (Waters Corporation, Billerica, MA, USA). TRIZAIC nanoTile 85 μm × 100 mm HSS-T3u wTRAP was used as separating device before mass spectrometer. Samples were loaded, trapped and washed for 2 min at 8.0 μl min−1 with 1% B. The analytical gradient used is as follows: 0–1 min 1% B; at 2 min 5% B; at 65 min 30% B; at 78 min 50% B; at 80 min 85% B; at 83 min 85% B; at 84 min 1% B; and at 90 min 1% B with 450 nl min−1. Buffers were made to ultra performance liquid chromatography-grade chemicals (Sigma-Aldrich); buffer A: 0.1% formic acid in water; and buffer B: 0.1% formic acid in acetonitrile.
The data were acquired in data-independent acquisition manner using HDMSE mode with Synapt G2-S HDMS (Waters Corporation). The HDMSE mode included ion mobility spectroscopy (IMS). The collected data range were 100–2000 m/z, scan time 1 s, IMS wave velocity 650 m s−1 and collision energy was ramped in trap between 20 and 60 V. Calibration was done with Glu1-fibrinopeptide B MS2 fragments and as a lock mass, Glu1-fibrinopeptide B precursor ion was used during the runs. The samples were run as triplicates, and further analysis was done with Progenesis QI for Proteomics software (Nonlinear Dynamics, Newcastle, UK).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD005263 (Vizcaino et al, 2016).
Data analysis
The data analysis of mass spectrometry data was performed as described previously (Saraswat et al, 2016). Briefly, the raw files were imported to Progenesis QI for proteomics software (Nonlinear Dynamics) using lock mass correction with 785.8426 m/z, corresponding to doubly charged Glu1-fibrinopeptide B. Default parameters for peak picking and alignment algorithm were used. The software facilitated the peptide identification with Protein Lynx Global Server and label-free quantification (Silva et al, 2006).
The peptide identification was done against Uniprot human FAST-All (FASTA) sequences (UniprotKB Release 2015_09, 20205 sequence entries) with (CLPB_ECOLI (P63285)), ClpB protein sequence inserted for label-free quantification. Modifications used were as follows: fixed at cysteine (carbamidomethyl) and variable in methionine (oxidation). Trypsin was used as digesting agent and one missed cleavage was allowed. Fragment and peptide error tolerances were set to auto and false discovery rate to <4%. One or more ion fragments per peptide, three or more fragments per protein and one or more peptides per protein were required for ion matching. These are default parameters in the software.
The identified proteins are grouped as one according to the parsimony principle, and also peptides unique to the protein are reported. Parsimony principle states that protein hits are reported as the minimum set that accounts for all observable peptides. Progenesis QI for proteomics does not take a strict parsimonious approach because of over-stringency as has been pointed out before (Serang et al, 2012). However, for resolution of conflicts, if two proteins contain some common peptides, protein with fewer peptides is subsumed into the protein with higher number of peptides, which are a superset of the subsumed protein’s peptides. All relevant proteins are listed as a group under the lead protein with greatest coverage or the highest score when the coverages of two or more proteins are equal. Quantitation is performed using the lead identity peptide data. More details about this approach can be accessed on the software website (www.nonlinear.com).
The proteins were considered different if they have a fold change 2 or more and an ANOVA P-value 0.05 or less. The ANOVA calculation assumes that the conditions are independent and applies the statistical test that assumes the means of the conditions are equal. The label-free protein quantitation was done with Hi-N method (Silva et al, 2006). In every injection the sample contained also 50 fmol of six CLPB_ECOLI (P63285, ClpB protein) peptides (Hi3 E. coli Standard, Waters). Hi3 peptides are used for normalising the peptide abundancies and relative quantitation was based on all the non-conflicting peptides found. The peptide ranking is done across all the runs. The abundancies of the peptides are averaged to provide a signal to the protein. Workings of the Progenesis softwares have been described in details on the software website (www.nonlinear.com) and also in published literature (Di Luca et al, 2015).
Differences between controls and cases were evaluated with ANOVA on a protein-to-protein basis. Principle component analysis was done with Progenesis QI for proteomics. EZinfo 3.0 (Release date 2 December 2014, Umetrics, Sweden) is a separate statistical package that can be used with Progenesis QI for proteomics. The data were imported into the EZinfo and supervised OPLS-DA modelling was performed, which gave us the variance vs correlation plot (S-Plot). Default parameters were used. Receiver-operating characteristic (ROC) curve analysis was also performed on some of the significantly different proteins predicted by S-Plot. Analyse-it programme, which works with Microsoft Excel, was used for making ROC curves with all the default parameters.
The unsupervised SOM-based clustering was also performed.
Further, unsupervised clustering was performed on centred and scaled expression values of same numbers of proteins using another clustering method – SOM, which is based on artificial neural models (Kohonen, 1997; Covell et al, 2003). The advantage of using SOM method is its efficiency in dealing with noisy and incomplete data in a robust manner. The SOM clustering was applied on protein expression data using som function in R with parameters (x-dim=5, y-dim=6, topology= ‘hexa’, neigh= ‘gaussian’). Heat map was finally generated to show the clustering using heatmap function in R.
Pathway analysis
Pathway analysis was performed as described previously (Saraswat et al, 2016). Briefly, Integrated Molecular Pathway Level Analysis (IMPaLA) was used for pathway overrepresentation analysis by their web-based service. The method and rationale behind this approach has been published previously (Kamburov et al, 2011). Ingenuity pathway analysis (IPA; Ingenuity Systems, Redwood City, CA, USA) was used for performing core analysis on the sperm cells proteomic data set with default parameters of the software. The results (canonical pathways) are presented in the Results section as a figure.
Results
Metadata
Thirteen serum samples from patients with OSCC together with 12 samples of healthy controls were included in this study, according to the criterion described in Materials and Methods. All cancers were cancers of the tongue and all patients except one were treated with curative intent. All the samples were collected in the operation theatre, before the start of the operation, that is, before resection of the cancer. The diagnosis for each patient had been confirmed by a head and neck pathologist preoperatively, by a tissue biopsy. None of the patients has received any treatment before the day of their surgery, on which the samples were taken. All the samples were fasting samples, and we did not find any correlation between smoking and protein expression profile. Patient and tumour demographics are presented in Supplementary Table 1. Every sample was run three times, and if there was dissimilarity between the triplicates in principal component analysis (PCA), the samples were excluded from further analysis. One such exclusion was done among the 13 OSCC samples (patient 1; Supplementary Table 1), leaving us with 12 OSCC samples.
Proteomics in cases and controls
In this protein analysis we identified 244 proteins with two or more unique peptides (Supplementary Table 2 and Figure 1). Fold change ranged from 65.19 to 1.01 when the highest mean was in cases and 4.12 to 1 when the highest mean was in controls. All protein abundancies in cases and controls were compared by ANOVA and the P-value ranged from 1.16 × 10−5 to 0.98. Altogether 165 proteins with two or more unique peptides had ANOVA P-value <0.05. (Supplementary Table 2 shows the protein list with two or more unique peptides and ANOVA P-value <0.05).

Data analysis workflow. MS, mass spectrometry. ANOVA: a test taking into account the mean difference, variance and the sample size was used. A general cutoff of.05 was used. PCA: analysis to determine and visualise the principal axes of protein abundance variation in cases and controls. The idea of using this biplot is to distinguish how much variation the classes of samples have when compared to one another and make it easily visible. OPLS-DA: a statistical method to find the predictive variance in comparison between cases and controls. OPLS-DA generates a S-Plot where x axis shows the magnitude of difference in particular protein abundance and y axis the significance of that protein in comparison of the two groups. A binary classification system called ROC analysis was used to further validate the S-Plot proteins by calculating AUC values. These predict which proteins selected by S-Plot also act as classifiers in ROC curve analysis. Clustering: serves as an alternative technique to analyse the difference between cases and controls. Self-organising maps (SOMs) is a data visualisation technique that reduces the dimensions of data through the use of self-organising neural networks. The results of SOM clustering methods are often visualised by a heat map. SOM belongs to unsupervised clustering. Pathway analysis: shows the enriched pathways among the proteins overexpressed in cases and controls. IPA and IMPaLA are two independent methods for this kind of analysis.
Principal component analysis
The PCA done in Progenesis QI proteomics was used to determine and visualise the principal axes of protein abundance variation in cases and controls. The PCA was performed first on all proteins with two or more unique peptides and it showed a partial tendency to separate cases and controls (Figure 2A). When PCA was done on proteins with two or more unique peptides and ANOVA P-value <0.05, the separation of the cases and controls was complete (Figure 2B). It is noteworthy, that ANOVA P-value, describing the inner variance of the groups, provides information only about the reliability of the fold change, and says anything about how high (or low) the fold change is. The two blue dots on the left in PCA space (Figure 2) represent two cases (patients 2 and 4; Table 1). Both have stage IV tumours, but based on the available clinical information, it is hard to say why they differ from other cases.

Principal component analysis using protein quantification data. (Upper panel) Principal component analysis (PCA) with two or more unique peptide-containing proteins. (Lower panel) PCA with two or more unique peptide-containing proteins passing the cutoff of 0.05 in ANOVA. The upper panel shows PCA done on proteins with two or more unique peptides, without any requirements for ANOVA P-value, the lower panel is done with ANOVA P-value <0.05. A trend towards separation between groups is seen in the first panel, and becomes complete on the second. Cases are marked with blue and controls with purple.
Orthogonal projections to latent structure-discriminant analysis
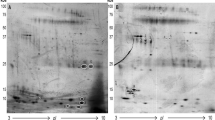
To model the differences between two groups OPLS-DA was used, and proteins that pass the cutoff value of ±0.65 for p(corr) values were considered significantly different between the two groups. An S-Plot was generated from the data (Figure 3).

S-plot of the OSCC cases vs healthy controls. S-plot obtained from OPLS-DA regression analysis. Proteins with p(corr) value higher than +0.65 or lower than −0.65 are considered significantly different. At the upper right corner are the proteins downregulated in the OSCC patients’ (cases) serum and at the lower left corner proteins upregulated in these cases.
The proteins obtained from S-Plot generated by OPLS-DA (Table 1) modelling were further analysed by ROC curve analysis. Analyse-it software (3.75, Analyse-it Software, Ltd., Leeds, UK) was used to calculate the area under the curve (AUC) values, with 95% confidence intervals (Table 1). Two out of the eight proteins had the AUC value of more than 0.9, considered excellent, and six had the AUC value of 0.81–0.89, considered good.
Clustering of the samples
Unsupervised clustering was performed on centred and scaled expression values of proteins using SOM. The SOM is a learning algorithm (Kohonen, 2012) based on artificial neural models. It clusters proteins with similar expression values to the same or nearby nodes. This clustering method gave us complete separation of cases (OSCC) and controls in the output (Figure 4). The separation was already complete at the level of all proteins considered (Supplementary Figure 1) and even better when only the proteins having significant values in ANOVA were considered (Figure 4).

The SOM heat map of the cases and controls. Heat map showing the complete separation of cases and controls in self-organising map (SOM) clustering when the proteins passing the cutoff of 0.05 in ANOVA were considered.
Pathway analysis
Two different tools were used for pathway analysis: IMPaLA (Kamburov et al, 2011); and IPA (Figure 5). Pathway overrepresentation analysis was performed using IMPaLA and the results are summarised in Supplementary Table 3. In addition, we created a Venn diagram showing the putative overlap of the enriched pathways among the proteins of highest means in cases and highest means in controls (Supplementary Figure 2). The protein lists were chosen by using ANOVA P-value <0.05. The Venn diagram shows that about a half of the pathways were unique to the controls, about one-fourth was common between cases and controls and another fourth unique to the cases.

Ingenuity IPA canonical pathways of all proteins with two or more unique peptides and ANOVA <0.05. Ingenuity pathway analysis ‘Core analysis’-enriched top canonical pathways are shown here. The IPA z-score scale predicts the activation (or inhibition) state of the respective signalling pathways. Horizontal axis is the –log (P-value) and vertical axis the given pathway. Blue, negative z-score; orange, positive z-score; white, z-score is 0; grey, no activity pattern available. The ratio of list genes found in each pathway over the total number of annotated genes into that pathway is shown as orange square. The orange threshold line corresponds to a P-value of 0.05.
The other pathway analysis tool included in the analyses was IPA, which showed numerous canonical pathways enriched in the data set. Only the top pathways are shown (Figure 5). In addition to acute phase response signalling, coagulation system and complement system, all tightly linked with tumourigenesis, liver X receptor and Farnesoid X receptor activation systems regulating lipid and cholesterol metabolism, clathrin-mediated endocytosis signalling and MSP–RON signalling pathways were among the enriched pathways.
Discussion
Head and neck squamous cell carcinoma accounts for more than half a million cases annually worldwide, and almost half of these patients will succumb to their disease in the first 5 years (Shen et al, 2015). Most HNSCCs leading to death are diagnosed in a clinically advanced stage and thus early detection of these tumours would be vital in trying to improve the prognosis and survival (Dahiya and Dhankhar, 2016). With the methods in use, only one-third of all HNSCC are diagnosed at an early stage (Dahiya and Dhankhar, 2016). Any findings of usable blood biomarkers would bring improvement to the current situation.
In our pilot study, we analysed the quantitative serum proteomics profiles of 13 patients with confirmed OSCC and 12 healthy control individuals from blood bank (Blood service). After depletion of the top 12 proteins to decrease the dynamic range, 244 proteins were quantified with two or more unique peptides. All the group classification methods consistently showed the two groups to separate from one another. Pathway analysis based on the protein levels showed significantly different pathways to be overrepresented in the two groups.
The OPLS-DA modelling and the subsequent S-Plot generated a list of most significant proteins in terms of separating the two groups (cases and controls) based on predictive variance (Figure 3 and Table 1). This is a powerful method to classify the groups of samples, resulting in outputs difficult to be extracted by other types of analyses. Proteins found to be significantly different among the two groups by S-Plot included eight proteins some of which were upregulated in OSCC serum samples while the others were downregulated. Among the proteins upregulated in OSCC were keratin type I and II, lactate dehydrogenase B chain. Further, the proteins downregulated in the serum of OSCC patients included platelet factor 4 and neutrophil defensin 1 among others (Table 1 for full details).
Lactate dehydrogenase B chain had the highest p(corr) score of 0.77 followed by other proteins described above. Only the proteins that pass the cutoff of +0.65 or −0.65 are presented in Table 1 and will be discussed here. Lactate dehydrogenase B chain is known to be expressed in blood and salivary gland and oesophageal regions. This protein is a key regulator of glycolysis, which is used as a primary source of energy by cancer cells. Inhibition of this protein, in gastric cancer, results in time- and dose-dependent inhibition of viability of cancer cells (Zhao et al, 2015). High expression of this protein in the cancer tissue has been found to be associated with poor overall survival and disease-free survival in OSCC (Sun et al, 2015). Other upregulated proteins in OSCC patients’ serum were keratin type I (keratin 32) and II (keratin 84). Keratins have an essential role in maintaining the structural integrity of healthy epithelium but have recently been shown to actively regulate also epithelial tumourigenesis (Karantza, 2011). There is evidence for the expression of both of these proteins in oral cavity and whole blood (Gene Cards, GCID:GC17M041460 and GCID:GC12M052377). Expression profiles of keratins in blood have been suggested to serve as potential biomarkers for different cancers such as OSCC, bladder cancer and small cell lung cancer (Lee et al, 2012; Hsu et al, 2015; Kuang et al, 2015). Among the upregulated protein in OSCC serum is another S-Plot significant protein, which can classify the OSCC and healthy serum, named as ribonuclease pancreatic (RNASE1). The protein RNASE1 is known to be elevated in serum of pancreatic cancer patients (Reddi and Holland, 1976). Another RNASE protein RNASEL was found to be mutated in HNC, breast and uterine cervix cancer and this mutation predicted the increased risk (Madsen et al, 2008). In addition to upregulation, also downregulation of genes as well as suppression of pathways is equally important for tumour growth and development. Downregulated proteins in OSCC patients' serum, compared to that of healthy controls, included platelet factor 4 and neutrophil defensin 1 among others.
De-regulation of defensins has been reported not only in inflammatory diseases but also in several cancers (Droin et al, 2009). Both tumour-promoting and -suppressing actions have been described – depending on, for example, the tumour type and cells expressing it (immune cells vs cancer cells; Droin et al, 2009). Defensin overexpression in OSCC tissue has also been reported (Lundy et al, 2004). However, this discrepancy may be due to the fact that most neutrophils will infiltrate the cancer tissue, and there are simply not many available to sustain levels of neutrophil defensins in blood. Moreover, most cancer patients develop neutropenia (Lustberg, 2012), which might explain the low levels of neutrophil defensin 1 in serum in our study. Platelet factor 4 supports platelet aggregation and is a key player in the coagulation homeostasis (Gerotziafas et al, 2001). Oncolytic viruses armed with platelet factor 4 were able to enhance the antiangiogenic effect and showed efficacy against tumours (Liu et al, 2006). It makes a lot of sense that HNC serum has low levels of this factor as tumours need angiogenesis to survive and proliferate.
Combining the data from both up- and downregulated proteins in OSCC patient’s serum enables us to form a picture of a dynamic disease with several altering incidents. Instead of seeking for just one protein with altered abundance in cancer patients’ serum, taking together a panel of proteins with modified expressions could form a more reliable composition for decision making, for example, in the future diagnostics of OSCC. In addition, a panel of proteins, such as those described by S-Plot in our study (Table 1), might be a better strategy than any one protein because cancers have individual specific changes also. Choosing only one protein biomarker might not work in all the patients, universally, of different age groups, ethnicity and tumour heterogeneity.
Statistical analysis and validation of high-throughput data is essential because sometimes fold change is not a good strategy, which surprisingly some studies still rely on. We have used ANOVA, OPLS-DA-based S-Plot, SOM clustering and ROC AUC values to establish individual protein’s validity to be used as biomarkers. Sometimes the fold changes can be high due to one or few outliers. Strict statistical validation helps to recognise these outliers. If a sample behaves as complete outlier on a global scale then PCA can immediately show it on the biplot space. Further we have used ANOVA, which can be used to crosscheck whether the fold changes of a given protein in case vs control also reflect reliable changes. For example, in our study, complement factor H-related protein 3 was found to be upregulated in OSCC samples with 3.27-fold change however the ANOVA values were very high and insignificant (0.0745). Upon careful inspection of the data from individual persons it showed that only a few patients had high values of this protein while others showed great variability. To check whether our proteomic analysis can be used to classify the samples into two groups by unsupervised method, SOM clustering was performed and samples were easily classified into two distinct groups (OSCC and controls; Figure 4). OPLS-DA S-Plot, which is a supervised method, was used to select the main protein features, which enable the classification of samples into two groups. Eight proteins described above were found and four of these proteins were also the ones that enabled the clustering of samples into two distinct groups in SOM clustering. It validates the significance of these proteins as they are being deemed significant by both supervised and unsupervised methods. These eight S-Plot significant proteins also had excellent ANOVA values of 1.03 × 10−02 to 1.16 × 10−5. Furthermore, in ROC these proteins had excellent AUC values (Table 1). It also shows that there is a degree of cross-talk among these statistical methods and they can be used to validate each other’s findings. This gives us high confidence about our suggested panel of biomarkers and they can be used in further studies to establish their value as screening as well as detection biomarker panel for OSCC.
Combining the data from both up- and downregulated proteins in OSCC patient’s serum enables us to form a picture of a dynamic disease with several altering incidents. Instead of seeking for just one protein with altered abundance in cancer patients’ serum, taking together a setting of proteins with modified expressions could form a more reliable composition for decision making, for example, in the future diagnostics of OSCC.
The protein expression data were further analysed in order to gain information on altered biological processes and pathways in OSCC vs healthy controls. This was done by comparing the pathways enriched in top-most abundant proteins in cases and controls. Again, two different methods were used to probe the pathway from these lists of proteins to serve as each other’s validation. Immune system and more specifically complement system, coagulation system and acute phase response system activation were found to be overrepresented pathways in OSCC by both methods used. These are obvious and logical findings for a tumour and are reported earlier in several cancers (Falanga et al, 2013a,2013b; Pio et al, 2013). To identify the pathways unique to the cases, Venn diagram comparing the pathways overrepresented in cases and controls was made, and several pathways linked to complement signalling were found among the pathways unique to the cases. Another interesting finding was the overrepresentation of clathrin-mediated endocytosis pathway in cases, when using IPA. Increasing evidence exists that altered endocytosis is one crucial element in cancer cell population to regulate tumour initiation and development, and clathrin-mediated endocytosis is one of the main mechanisms of endocytosis (Mosesson et al, 2008).
In IPA analysis, RON–MS signalling pathway was overrepresented in OSCC patient’s serum. The RON is a proto-oncogene present in multiple different epithelial cell types, malignant and non-malignant (Gaudino et al, 1995; Iwama et al, 1995). Its ligand is called macrophage-stimulating hormone (MSP), belonging to the plasminogen–prothrombin gene family (Camp et al, 2005). The activation of RON by MSP leads to the activation of several pathways resulting in tumour progression and metastasis, through cell adhesion, invasion, motility and proliferation together with inhibited apoptosis (Camp et al, 2005). A few studies of RON–MSP signalling in HNC exist, reporting RON expression to be linked with both higher levels of epidermal growth factor receptor and poorer disease specific survival (Lin et al, 2004; Keller et al, 2013; Yoon et al, 2013). Epidermal growth factor receptor inhibitors are used in the treatment of HNSCC, although the results are somehow unpredictable and not that marked (Keller et al, 2013). A picture emerges from these results that these tumours are complex and involve multiple altering events in protein expression and associated pathways, which is in accordance with current knowledge.
To conclude, a total of 244 proteins were quantified with two or more peptides in the serum samples of OSCC patients and their healthy controls. Both supervised and unsupervised methods were used for group classification and they showed consistent separation of these two groups. By pathway analysis done by two independent methods we were able to identify the biological processes altered in OSCC when comparing with those of healthy controls. The idea of this kind of discovery-driven, system-level analysis of a disease is to provide novel high-confidence targets for more in-depth studies.
We identified a set of eight proteins selected by S-Plot that serves as a potential panel of biomarkers, separating the OSCC patients from healthy individuals in our pilot study. It would be of very high value to screen this panel of biomarkers in a larger cohort of patients to establish their routine clinical use.
Change history
25 July 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bagan JV, Scully C (2008) Recent advances in Oral Oncology 2007: epidemiology, aetiopathogenesis, diagnosis and prognostication. Oral Oncol 44 (2): 103–108.
Braakhuis BJ, Leemans CR, Visser O (2014) Incidence and survival trends of head and neck squamous cell carcinoma in the Netherlands between 1989 and 2011. Oral Oncol 50 (7): 670–675.
Camp ER, Liu W, Fan F, Yang A, Somcio R, Ellis LM (2005) RON, a tyrosine kinase receptor involved in tumour progression and metastasis. Ann Surg Oncol 12 (4): 273–281.
Chaturvedi AK, Anderson WF, Lortet-Tieulent J, Curado MP, Ferlay J, Franceschi S, Rosenberg PS, Bray F, Gillison ML (2013) Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol 31 (36): 4550–4559.
Covell DG, Wallqvist A, Rabow AA, Thanki N (2003) Molecular classification of cancer: unsupervised self-organizing map analysis of gene expression microarray data. Mol Cancer Ther 2 (3): 317–332.
Dahiya K, Dhankhar R (2016) Updated overview of current biomarkers in head and neck carcinoma. World J Methodol 6 (1): 77–86.
Decker J, Goldstein JC (1982) Risk factors in head and neck cancer. N Engl J Med 306 (19): 1151–1155.
Di Luca A, Henry M, Meleady P, O’Connor R (2015) Label-free LC-MS analysis of HER2+ breast cancer cell line response to HER2 inhibitor treatment. Daru 23 (1): 40.
Droin N, Hendra JB, Ducoroy P, Solary E (2009) Human defensins as cancer biomarkers and antitumour molecules. J Proteomics 72 (6): 918–927.
Falanga A, Marchetti M, Vignoli A (2013a) Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 11 (2): 223–233.
Falanga A, Russo L, Verzeroli C (2013b) Mechanisms of thrombosis in cancer. Thromb Res 131 (Suppl 1): S59–S62.
Gaudino G, Avantaggiato V, Follenzi A, Acampora D, Simeone A, Comoglio PM (1995) The proto-oncogene RON is involved in development of epithelial, bone and neuro-endocrine tissues. Oncogene 11 (12): 2627–2637.
Gerotziafas GT, Elalamy I, Lecrubier C, Lebrazi J, Mirshahi M, Potevin F, Lecompte T, Samama MM (2001) The role of platelet factor 4 in platelet aggregation induced by the antibodies implicated in heparin-induced thrombocytopenia. Blood Coagul Fibrinolysis 12 (7): 511–520.
Hsu YP, Hsieh CH, Chien HT, Lai CH, Tsao CK, Liao CT, Kang CJ, Wang HM, Chang JT, Huang SF (2015) Serum markers of CYFRA 21-1 and C-reactive proteins in oral squamous cell carcinoma. World J Surg Oncol 13: 253.
Iwama A, Wang MH, Yamaguchi N, Ohno N, Okano K, Sudo T, Takeya M, Gervais F, Morissette C, Leonard EJ, Suda T (1995) Terminal differentiation of murine resident peritoneal macrophages is characterized by expression of the STK protein tyrosine kinase, a receptor for macrophage-stimulating protein. Blood 86 (9): 3394–3403.
Kamburov A, Cavill R, Ebbels TMD, Herwig R, Keun HC (2011) Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 27 (20): 2917–2918.
Karantza V (2011) Keratins in health and cancer: more than mere epithelial cell markers. Oncogene 30 (2): 127–138.
Keller J, Nimnual AS, Shroyer KR, Joy C, Ischenko I, Chandler CS, Dong LM, Hayman MJ, Chan EL (2013) Ron tyrosine kinase receptor synergises with EGFR to confer adverse features in head and neck squamous cell carcinoma. Br J Cancer 109 (2): 482–492.
Kohonen T (1997) Self-organizing Maps Vol. 117. Springer: Berlin, Germany.
Kohonen T (2012) Self-organizing Maps. Springer: Berlin, Germany.
Kuang LI, Song WJ, Qing HM, Yan S, Song FL (2015) CYFRA21-1 levels could be a biomarker for bladder cancer: a meta-analysis. Genet Mol Res 14 (2): 3921–3931.
Kuusela P, Saraswat M, Joenväärä S, Kaartinen J, Järvinen A, Renkonen R (2017) Changes in plasma protein levels as an early indication of a bloodstream infection. PLoS One 12 (2): e0172987.
Lee HJ, Kim YT, Park PJ, Shin YS, Kang KN, Kim Y, Kim CW (2012) A novel detection method of non-small cell lung cancer using multiplexed bead-based serum biomarker profiling. J Thorac Cardiovasc Surg 143 (2): 421–427.
Lin HS, Berry GJ, Fee WE Jr, Terris DJ, Sun Z (2004) Identification of tyrosine kinases overexpressed in head and neck cancer. Arch Otolaryngol Head Neck Surg 130 (3): 311–316.
Liu T-C, Zhang T, Fukuhara H, Kuroda T, Todo T, Martuza RL, Rabkin SD, Kurtz A (2006) Oncolytic HSV armed with platelet factor 4, an antiangiogenic agent, shows enhanced efficacy. Mol Ther 14 (6): 789–797.
Lundy FT, Orr DF, Gallagher JR, Maxwell P, Shaw C, Napier SS, Gerald Cowan C, Lamey PJ, Marley JJ (2004) Identification and overexpression of human neutrophil alpha-defensins (human neutrophil peptides 1, 2 and 3) in squamous cell carcinomas of the human tongue. Oral Oncol 40 (2): 139–144.
Lustberg MB (2012) Management of neutropenia in cancer patients. Clin Adv Hematol Oncol 10 (12): 825–826.
Machiels JP, Lambrecht M, Hanin FX, Duprez T, Gregoire V, Schmitz S, Hamoir M (2014) Advances in the management of squamous cell carcinoma of the head and neck. F1000Prime Rep 6: 44.
Madsen BE, Ramos EM, Boulard M, Duda K, Overgaard J, Nordsmark M, Wiuf C, Hansen LL (2008) Germline mutation in RNASEL predicts increased risk of head and neck, uterine cervix and breast cancer. PLoS One 3 (6): e2492.
Mosesson Y, Mills GB, Yarden Y (2008) Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer 8 (11): 835–850.
Pio R, Ajona D, Lambris JD (2013) Complement inhibition in cancer therapy. Semin Immunol 25 (1): 54–64.
Razzouk S (2014) Translational genomics and head and neck cancer: toward precision medicine. Clin Genet 86 (5): 412–421.
Reddi KK, Holland JF (1976) Elevated serum ribonuclease in patients with pancreatic cancer. Proc Natl Acad Sci USA 73 (7): 2308–2310.
Saraswat M, Joenväärä S, Jain T, Tomar AK, Sinha A, Singh S, Yadav S, Renkonen R (2016) Human spermatozoa quantitative proteomic signature classifies normo- and asthenozoospermia. Mol Cell Proteomics 16 (1): 57–72.
Serang O, Moruz L, Hoopmann MR, Käll L (2012) Recognizing uncertainty increases robustness and reproducibility of mass spectrometry-based protein inferences. J Proteome Res 11 (12): 5586–5591.
Shen W, Sakamoto N, Yang L (2015) Cancer-specific mortality and competing mortality in patients with head and neck squamous cell carcinoma: a competing risk analysis. Ann Surg Oncol 22 (1): 264–271.
Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel ms acquisition. Mol Cell Proteomics 5 (1): 144–156.
Smith EM (1979) Epidemiology of oral and pharyngeal cancers in the United States: review of recent literature. J Natl Cancer Inst 63 (5): 1189–1198.
Sun W, Zhang X, Ding X, Li H, Geng M, Xie Z, Wu H, Huang M (2015) Lactate dehydrogenase B is associated with the response to neoadjuvant chemotherapy in oral squamous cell carcinoma. PLoS One 10 (5): e0125976.
van Dijk BA, Brands MT, Geurts SM, Merkx MA, Roodenburg JL (2016) Trends in oral cavity cancer incidence, mortality, survival and treatment in the Netherlands. Int J Cancer 139 (3): 574–583.
Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44 (D1): D447–D456.
Wynder EL, Mushinski MH, Spivak JC (1977) Tobacco and alcohol consumption in relation to the development of multiple primary cancers. Cancer 40 (4 Suppl): 1872–1878.
Yoon TM, Kim SA, Park YL, Lee KH, Sung MW, Lee JK, Lim SC, Chung IJ, Joo YE (2013) Expression of the receptor tyrosine kinase recepteur d'origine nantais and its association with tumour progression in hypopharyngeal cancer. Head Neck 35 (8): 1106–1113.
Zhao Z, Han F, Yang S, Wu J, Zhan W (2015) Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett 358 (1): 17–26.
Acknowledgements
This study was supported by Korvatautien tutkimussäätiö and Jane and Aatos Erkko foundation.
Author contributions
MS: prepared the samples, performed data analysis and participated in writing the manuscript; AM: participated in writing the paper and study design; RA: SOM analysis and clustering; SJ: mass spectrometry and data analysis; SR: design of the study and writing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Saraswat, M., Mäkitie, A., Agarwal, R. et al. Oral squamous cell carcinoma patients can be differentiated from healthy individuals with label-free serum proteomics. Br J Cancer 117, 376–384 (2017). https://doi.org/10.1038/bjc.2017.172
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.172
Keywords
This article is cited by
-
A systematic review of proteomic biomarkers in oral squamous cell cancer
World Journal of Surgical Oncology (2021)
-
How I Approach Laryngopharyngoesophageal Reflux (LPR)
Current Gastroenterology Reports (2021)
-
Label-free serum proteomics and multivariate data analysis identifies biomarkers and expression trends that differentiate Intraductal papillary mucinous neoplasia from pancreatic adenocarcinoma and healthy controls
Translational Medicine Communications (2019)
-
Thromboinflammatory changes in plasma proteome of pregnant women with PCOS detected by quantitative label-free proteomics
Scientific Reports (2019)
-
Patients with early-stage oropharyngeal cancer can be identified with label-free serum proteomics
British Journal of Cancer (2018)

{kind=link}
{kind=link}