
Abstract
The p53 family of transcription factors is essential to counteract tumour formation and progression. Although previously this was exclusively associated with the ability of the p53 family to induce cell cycle arrest and apoptosis, an increasing number of reports have now indisputably demonstrated that the tumour suppressive functions of the p53 family members also rely on their ability to control and regulate cellular metabolism and maintain cellular oxidative homeostasis. Here, we review how each p53 family member, including p63 and p73, controls metabolic pathways in physiological conditions, and how these mechanisms could be exploited to provide anticancer therapeutic opportunities.
Similar content being viewed by others

Main
The p53 (TP53) gene is the most frequently mutated gene in human cancers (Kandoth et al, 2013), and has consequently captivated the attention of the cancer research community and is one of the most studied and best characterised tumour suppressor genes. This interest has been nourished by the phenotypes shown by numerous mouse models, including p53 knockout mice, that spontaneously develop tumours (Donehower et al, 1992; Jacks et al, 1994), as well as mice carrying somatic p53 mutations, that phenocopy Li–Fraumeni syndrome (Lang et al, 2004; Olive et al, 2004), a condition predisposing to early onset of several tumour types.
Initial efforts made to unveil the mechanisms utilised by p53 to mediate tumour suppression highlighted its ability to be induced by a variety of intrinsic and extrinsic stimuli, such as DNA damage, oncogene activation, ribosomal stress, hypoxia, heat shock, and others. In turn, p53 activates the expression of a large network of genes involved in many cellular processes including cell cycle control and programmed cell death. During the past decade, however, our knowledge of the p53 transcriptome became more comprehensive because of the ease of conducting genome-wide analyses. These analyses further indicated that p53 is a master regulator of many other biological processes, such as autophagy, differentiation, and tumour microenvironment remodelling (Bieging et al, 2014). The importance of these additional mechanisms in mediating p53 tumour suppression was recently underlined by various mouse models demonstrating that p53 tumour suppressive activities are maintained even when its regulation of cell cycle and programmed cell death are impaired (Brady et al, 2011; Li et al, 2012; Timofeev et al, 2013; Valente et al, 2013). Notably, one of these mouse models showed that lack of p53 classical responses (i.e., cell cycle arrest, apoptosis, and senescence) does not affect its capacity to control the expression of metabolism-related genes, through which p53 regulates glucose uptake and catabolism, and inhibits the production of reactive oxygen species (ROS; Li et al, 2012). However, when additional mutations abolishing p53 regulation of metabolism-related genes are introduced, p53 antitumour functions are restrained (Wang et al, 2016), thereby indicating that metabolic control by p53 is crucial to drive tumour suppression. These results are in line with the numerous connections established over the years between metabolism-related stressors (such as imbalanced nucleotide pools or nutrient deprivation) and p53 classical responses (Linke et al, 1996; Agarwal et al, 1998; Hastak et al, 2008) that indicate that the various p53-regulated processes are not compartmentalised, and instead they can synergistically promote each other to ultimately maintain cellular homeostasis and counteract tumour formation and progression (Bieging et al, 2014).
In performing its tumour suppressive activities, p53 acts in concert with the other members of its family, p63 and p73. In response to genotoxic stress, both proteins are recruited to the promoters of some p53 target genes and are essential for the proper induction of genes involved in apoptosis by p53 (Flores et al, 2002), a property relying on the high homology of the DNA-binding domains of these three transcription factors. In addition to cooperating in the regulation of the same set of genes, each p53 family member has its own transcriptional repertoire, whose distinctiveness is ascribed, at least in part, to the structural differences of these proteins (Botchkarev and Flores, 2014). In particular, both the p63 (TP63) and the p73 (TP73) genes encode two sets of abundantly expressed isoforms: full-length isoforms (called TAp63 and TAp73) having a transactivation domain similar to that present in p53 (known as TA1), and N-terminal truncated isoforms (ΔNp63 and ΔNp73) that are devoid of that domain and instead present an alternative and shorter transactivation domain (known as TA2) (Duijf et al, 2002; Orzol et al, 2015). Compared with the amino terminal truncated isoforms of p53 (ΔN40, ΔN133, and ΔN160), whose specific activities are still under investigation (Joruiz and Bourdon, 2016), the different functions fulfilled by the many p63 and p73 isoforms have been elucidated by the isoform-specific knockout mouse models generated over the past decade. These mouse models have revealed that both TAp63 and TAp73 are potent tumour suppressor genes: lack of either gene leads to spontaneous development of tumours in mice (Tomasini et al, 2008; Su et al, 2010). Intriguingly, their tumour suppressive activities are associated with the ability of these proteins to control lipid and glucose metabolism and mitochondrial function, respectively (Rufini et al, 2012; Su et al, 2012). On the other hand, ΔNp63 and ΔNp73 knockout mice have developmental defects in the epidermis and limbs, and in the nervous system, respectively (Wilhelm et al, 2010; Chakravarti et al, 2014; Restelli et al, 2014). Both proteins act as dominant negative by binding to the other members of the family and inhibiting their functions. A detailed mechanism underlying the oncogenic properties of ΔNp63 and ΔNp73 was recently connected with their ability to regulate metabolic reprogramming in tumours (Venkatanarayan et al, 2015).
Given the numerous links between the p53 family members and metabolic pathways, here we review the physiological roles of these transcription factors in controlling cellular metabolism and discuss how these connections reverberate through tumour initiation and progression, thus providing opportunities that can be exploited for cancer treatment.
The p53 family and carbohydrate metabolism
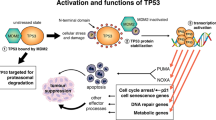
Glucose is one of the primary energy sources for both normal and cancer cells and the p53 family controls its metabolism at multiple levels (Figure 1). First, both p53 and TAp63 regulate the balance of glucose uptake via the many glucose transporters present on the cytoplasmic membrane. In the case of high intracellular levels of glucose, p53 directly represses the expression of GLUT-1 and GLUT-4 (Schwartzenberg-Bar-Yoseph et al, 2004), and it decreases expression of GLUT-3 by antagonising the NF-κB pathway (Kawauchi et al, 2008). On the contrary, when intracellular levels of glucose are low, TAp63 promotes glucose uptake, even though the involved glucose transporters are not currently defined (Su et al, 2012). Once internalised, glucose is catabolised through glycolysis, whose first step is mediated by hexokinase II, encoded by the p53 transcriptional target gene HK2 (Mathupala et al, 1997). The substrate of hexokinase II activity, glucose-6-phospate (G6P) can fuel two alternative pathways: glycolysis and the pentose phosphate pathway (PPP). In human and mouse cell lines, both in physiological conditions and in response to oxidative stress, p53 inhibits subsequent steps of the glycolysis pathway through its transcriptional target genes, TIGAR (Bensaad et al, 2006) and miR-34a (Kim et al, 2013), thereby shunting G6P towards the PPP. This pathway generates several metabolites, including precursors of nucleotides and aromatic amino acids, and allows for the production of reducing equivalents in the form of NADPH that in turn supports p53 regulation of the cellular oxidative homeostasis (Bensaad et al, 2006). TIGAR promotes the PPP and the formation of NAPDH through the elimination of ROS as demonstrated in TIGAR−/− mice that, although developmentally normal, are characterised by defects in ROS scavenging capacity and reduced tissue regeneration potential (Cheung et al, 2013).

Crosstalk between the p53 family members and metabolic pathways. The p53 (pink) and its family members, TAp63 (orange) and TAp73 (blue), control glucose uptake and inhibit glycolysis while enhancing fatty acid oxidation and favouring mitochondrial maintenance and activity.
The PPP flux is also enhanced by TAp73 through the induction of glucose-6-phosphate dehydrogenase (G6PD), the first and rate-limiting enzyme of the PPP (Jiang et al, 2013b). In human cancer cell lines, instead, p53 inhibits the PPP pathway by secluding G6PD (Jiang et al, 2011), thus depleting cells of antioxidants and leading to excessive oxidative stress and cell death. Independently of p53, both TAp63 and TAp73 can achieve the same effect through the induction of IAPP (Venkatanarayan et al, 2015); its gene product amylin is an inhibitor of hexokinase II preventing G6P formation and utilisation for the PPP. The relevance of amylin’s tumour suppressive and preventive activity was demonstrated by its synthetic analogue pramlintide, an FDA approved antidiabetic drug that leads to tumour regression in p53−/− mice (Venkatanarayan et al, 2015, 2016).
The involvement of the p53 family in carbohydrate metabolism is also crucial during the final steps of glycolysis (Figure 1). The by-product of this pathway, lactate, is generally expelled by cells through the monocarboxylate transporter 1 (MCT1), whose expression is repressed by p53 in hypoxic conditions (Boidot et al, 2012). The subsequent accumulation of lactate prevents glycolysis to further take place and forces its precursor pyruvate to feed the tricarboxylic acid (TCA) cycle, an event additionally promoted by p53 that sustains the conversion of pyruvate into acetyl-CoA (Contractor and Harris, 2012). Concurrently, p53 inhibits pyruvate recycling by the TCA cycle through the repression of the two malic enzymes ME1 and ME2 (Jiang et al, 2013a) and provides additional fuel for this pathway by inducing GSL2 (Hu et al, 2010; Suzuki et al, 2010), thus overall increasing the TCA cycle rate. Mass spectrometry experiments performed upon overexpression of TAp73 suggested that it supports the TCA cycle by increasing the levels of several enzymes involved in this pathway (D'Alessandro et al, 2013). However, further investigation is needed to unveil the connections of the TCA cycle with TAp73 at physiological levels, as well as with the other members of the p53 family.
Overall, the p53 family members, and in particular p53 and TAp63, cooperate in regulating intracellular glucose levels by balancing glucose uptake accordingly, and promote the diversion of glucose metabolites towards PPP by acting on different steps of the glycolytic pathway.
Lipid metabolism control by the p53 family
Lipids have the highest caloric content among the biological macromolecules and represent the best energetic storage for cells; therefore, both fatty acid catabolism and anabolism are tightly regulated and kept in check by the p53 family. Synthesis of fatty acids is restricted to specialised tissues, including adipose tissue, liver, and lactating mammary glands. Both p53 and TAp63 concur in inhibiting this process as highlighted by the proclivity of their respective knockout mouse models to become obese (Su et al, 2012; Wang et al, 2013). In the case of p53, the counteraction of lipid biogenesis is mainly achieved in the adipocytes through the repression of the master regulator of fatty acid synthesis, SREBP1, that in turn cannot trigger the expression of several enzymes involved in this pathway, including acetyl-CoA carboxylase (ACC), ATP citrate lyase (ACLY), and fatty acid synthase (FASN) (Yahagi et al, 2003). TAp63 also controls downregulation of FASN and of other lipogenic enzymes through the concerted transcriptional activation of AMPKα2, LKB1 (also known as STK11), and SIRT1 (Su et al, 2012). This TAp63-mediated inhibition of lipid anabolism can be supported by metformin, a drug widely used to treat type II diabetes and reported to increase TAp63 levels in order to accomplish its antidiabetic effects (Su et al, 2012). A contribution opposite to p53 and TAp63 is provided by ΔNp63, whose induction of FASN in head and neck squamous cell carcinomas is required for ΔNp63-dependent prosurvival activities (Sabbisetti et al, 2009).
Mevalonate (MVA) is another important lipogenic pathway that allows for the synthesis of cholesterol. Many of the enzymes involved in the MVA pathway are induced by p53 that in this way can control both membrane permeability and the synthesis of cholesterol-derived hormones (Laezza et al, 2015). In tumours harbouring p53 mutations, these proteins interact with SREBP1 thus hijacking the MVA pathway and promoting the mutant p53-associated aggressiveness of several breast cancers (Freed-Pastor et al, 2012). This phenomenon can be counteracted by the utilisation of clinically available HMG-CoA reductase inhibitors, such as simvastatin (Freed-Pastor et al, 2012), that could represent a valid therapeutic opportunity to treat mutant p53-addicted tumours. As many of the gain-of-function properties of mutant p53 rely on its ability to inhibit TAp63 and TAp73 (Walerych et al, 2012), it would be interesting to investigate whether these two p53 family members may also affect the MVA pathway.
Lipid anabolism needs to be coordinated and compensated by fatty acid oxidation (FAO) that enables lipid stored energy to be utilised. In opposition to the limited number of tissues where de novo fatty acid biosynthesis takes place, this mitochondrial catabolic pathway occurs in all the cells of the body apart from red blood cells and neurons. The FAO mediates the degradation of fatty acids into two-carbon moieties that – once channelled into acetyl-CoA – may fuel the TCA cycle. The first step of FAO, that is the translocation of the fatty acids into the mitochondria, is performed by carnitine acetyltransferases, induced by both p53 (Sanchez-Macedo et al, 2013) and TAp63 (Su et al, 2012; Figure 1). In general, both p53 family members cooperate in reducing lipid synthesis and promoting lipid degradation, thus counteracting predisposition to obesity and tumour formation (Su et al, 2013). The other member of the family, p73, is also implicated in the regulation of lipid metabolism. The p73 knockout mice show abnormal lipid accumulation in the liver because of impaired triglyceride hydrolysis, with in vitro data pointing at an involvement of TAp73 in such a process (He et al, 2013, 2015). It would be interesting to assess the in vivo role of each p73 isoform, namely TAp73 and ΔNp73, in lipid metabolism in the respective isoform-specific knockout mice, even though a possible lack of evidence could be in line with some of the distinctive functions of these isoforms compared with the rest of their family (Napoli and Flores, 2016).
The p53 family dictates mitochondrial activities and ros production
The coordinated regulation of glycolysis and FAO by p53 and TAp63 causes the channelling of both pathways in the synthesis of acetyl-CoA, thus promoting the TCA cycle and the production of NADH and FADH2. These reducing agents are then exploited by the oxidative phosphorylation (OXPHOS) in the mitochondria to complete cellular respiration. The integration of these pathways allows the most efficient generation of ATP molecules per amount of glucose. In cancer cells, however, such a concerted action does not always occur and glucose may be entirely processed into lactate through glycolysis even in normoxic conditions. This was one of the earliest metabolic features of cancer cells to be discovered and is now known as Warburg effect (Warburg, 1925).
In addition, p53 sustains OXPHOS by inducing the expression of enzymes of the mitochondrial electron transport chain, including cytochrome c oxidase 2 (SCO2; Matoba et al, 2006) and the mitochondrial encoded cytochrome c oxidase 1 (MT-CO1) (Okamura et al, 1999). The latter observation is in agreement with the broader activity of p53 in mitochondrial maintenance. Indeed, p53 can localise to the mitochondria (Marchenko et al, 2000), where it controls mitochondrial genomic integrity through its interaction with mitochondrial DNA polymerase γ (Achanta et al, 2005). Furthermore, p53 regulates mitochondrial DNA copy number and mitochondrial mass through the induction of the p53-controlled ribonucleotide reductase (p53R2; Bourdon et al, 2007), and it is required for the proper removal of damaged mitochondria (i.e., mitophagy) via the upregulation of the mitochondria-eating protein (Mieap; Kitamura et al, 2011).
Although p53 and TAp63 promote OXPHOS at multiple levels, the support by TAp73 mainly relies on its ability to induce the expression of cytochrome c oxidase 4 isoform 1 (Cox4i1; Flores and Lozano, 2012; Rufini et al, 2012; Figure 1). As reported for the other family members (Su et al, 2009, 2012; Vigneron and Vousden, 2010), altered mitochondrial functions due to loss of TAp73 decrease oxygen consumption and ATP production by the mitochondria that in turn are associated with increased oxidative damage and senescence in vitro, and premature ageing in vivo (Rufini et al, 2012). All these effects are attributable to the increased levels of ROS, whose intracellular amounts are strictly regulated by all the p53 family members. p53 has been reported to both reduce and increase ROS levels, and this Janus effect is associated with different cellular conditions. In physiological conditions as well as under minor metabolic stress, p53 participates in reducing ROS levels through multiple approaches, including: (1) maintenance of mitochondrial integrity (Park et al, 2016); (2) induction of TIGAR that stimulates NADPH production by the PPP pathway (Bensaad et al, 2006); (3) promotion of GSH synthesis after serine depravation (Maddocks et al, 2013); (4) upregulation of several antioxidant factors, such as aldehyde dehydrogenase 4 (ALDH4; Yoon et al, 2004), sestrin-1 and -2 (SESN1 and SESN2) (Budanov and Karin, 2008), and tumour protein p53 inducible nuclear protein 1 (TP53INP1; Cano et al, 2009); and (5) repression of pro-oxidant genes like cyclooxygenase 2 (COX2; Subbaramaiah et al, 1999) and nitric oxide synthase (NOS; Ambs et al, 1998). However, in the case of severe damage, p53 increases ROS levels to eliminate the damaged cells through both induction of pro-oxidative genes (Zhuang et al, 2012) and inhibition of antioxidant genes, including G6PD (Jiang et al, 2011), ME1 and ME2 (Jiang et al, 2013a), and manganese superoxide dismutase (SOD2; Zhao et al, 2005). These p53-dependent augmented ROS levels can be further increased by the p53-mediated repression of PGC-1α and PGC-1β, two transcription factors required for mitochondrial biogenesis, in response to irreparable damage such as telomere shortening (Sahin et al, 2011). In p53-deficient or -mutated cancer cells, both TAp63 and TAp73 can substitute for p53 pro-oxidative functions by upregulating IAPP (Venkatanarayan et al, 2015) that inhibits hexokinase 2 and G6P formation to hinder PPP flux and the production of antioxidant equivalents. Hence, in physiological condition, p53, TAp63, and TAp73 cooperate in keeping ROS production under control, but, in line with their functions as tumour suppressors, these transcription factors can also exploit ROS to eliminate any overly damaged or cancerous cell.
Caloric restriction, longer lifespan, and the p53 family
A constantly growing body of compelling evidence obtained in multiple organisms clearly shows a correlation between restrained metabolism (i.e., caloric restriction) and increased lifespan (Ruetenik and Barrientos, 2015). Reduced availability of nutrients, such as glucose and amino acids, primarily triggers autophagy, a catabolic mechanism allowing protein recycle and degradation of damaged organelles (Napoli and Flores, 2013). This biological process can be activated by p53, TAp63, and TAp73 as stress response (Kenzelmann Broz et al, 2013), and it is essential to counteract ageing and age-related diseases (Martinez-Lopez et al, 2015). In line with this, one of the proposed physiological functions of p53 is to delay ageing. Indeed, transgenic mice expressing multiple copies of p53 under its endogenous promoter – therefore maintaining its normal regulation – are characterised by increased lifespan (Matheu et al, 2007). On the contrary, any alterations of the p53 physiological activity, either its inhibition through mutations (as in p53 S18A) or its stable hyperactivation possibly because of the expression of C-terminal fragments, correlate with reduced longevity, osteoporosis, and other signs of accelerated ageing (Tyner et al, 2002; Armata et al, 2007). The protective role in ageing of physiological p53 levels could be attributed to the multiple connections between p53 and the two nutrient sensors and autophagy modulators, mTOR and Sirt1 (Tucci, 2012; Napoli and Flores, 2013). Both factors are crucial regulators of ageing. The mTOR is the hub on which nutrient levels and growth factor signalling pathways converge to block autophagy; therefore, either pharmacological or mutational inhibition of the mTOR pathway can increase longevity in a variety of in vivo models, ranging from yeast to mice (Johnson et al, 2013). Sirt1 can function as an indirect sensor of the cellular oxidative state and it activates autophagy in response to oxidative stress detected as high NAD+ levels. Although the relevance of Sirt1 in expanding lifespan may vary according to the considered animal model and could be concealed by the presence of other sirtuins having redundant function (Ramis et al, 2015), Sirt1-overexpressing mice show reduced incidence of age-related metabolic disorders such as diabetes and liver steatosis (Banks et al, 2008). Intriguingly, Sirt1 is a direct TAp63 target gene (Su et al, 2012) and TAp63−/− mice, which have low Sirt1 levels, are characterised both by impaired glucose and lipid metabolism (Su et al, 2012) and by premature ageing and reduced lifespan (Su et al, 2009). Similar reduced lifespan is observed in TAp73−/− mice and, also in this case, the defect is associated with metabolic dysfunctions (Rufini et al, 2012).
Taken together, these in vivo mouse models underline the crucial connections between metabolic pathways, ageing, and the tumour suppressive members of the p53 family.
Targeting metabolic pathways as anticancer strategy
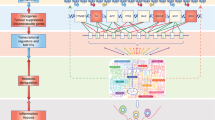
The numerous connections interweaving the p53 family members with the above-discussed metabolic pathways are present in both normal and cancer cells. As a consequence, several compounds able to interfere with either glucose or lipid metabolism have been demonstrated to sustain the tumour suppressive activities of p53, TAp63, and TAp73 (Figure 2). One of the best-investigated examples is metformin. For more than a decade, reports have been accumulating regarding the antineoplastic properties of this drug that is primarily used for its antidiabetic efficacy (Aldea et al, 2014). Intriguingly, part of this anticancer effect can be attributed to the capability of metformin to increase the levels of TAp63, in turn promoting the TAp63-mediated induction of genes crucial for both metabolic regulation and tumour suppression, such as AMPKα2, LKB1, and SIRT1 (Su et al, 2012). Another promising anticancer strategy is represented by the usage of statins (Matusewicz et al, 2015) that have been proved to limit the oncogenic properties of mutant p53 (Freed-Pastor et al, 2012). Given that one of the main features of mutant p53 is to sequester TAp63 and TAp73, it can be hypothesised that part of the tumour suppressive activities associated with statins might be achieved by unleashing these two transcription factors. In addition to inhibition by mutant p53, TAp63 and TAp73 can also be bound and blocked by ΔNp63 and ΔNp73 (Orzol et al, 2015). It has recently been demonstrated that ΔNp63 can be targeted by HDAC inhibitors (HDACi) that efficaciously reduce the levels of ΔNp63 through Fbw7 (Napoli et al, 2016). As tumour regression associated with loss of ΔNp63 is also accompanied by metabolic reprogramming (Venkatanarayan et al, 2015), it would be interesting to verify whether the therapeutic activity of HDACi may partially rely on alterations in metabolism caused by the reactivation of TAp63 and TAp73. Once these two transcription factors are released from the inhibition of a dominant negative member of the family (namely mutant p53, ΔNp63, or ΔNp73), they induce the expression of IAPP, whose synthetic analogue, pramlintide, was proven to act as a potent anticancer drug in preclinical models (Venkatanarayan et al, 2015, 2016).

The FDA-approved drugs that enhance p53 family tumour suppression through metabolic reprogramming.The FDA-approved compounds promote the transcriptional activity of TAp63 (orange) and TAp73 (blue), ultimately supporting the p53 family-mediated tumour suppression through transcriptional regulation of metabolic reprogramming.
Taken together, these different classes of molecules (metformin, statins, HDACi, and pramlintide) indicate that perturbing the p53 family to achieve metabolic reprogramming can be an effective strategy to counteract tumour formation and progression. Hence, we deem that this evidence should prompt the investigation of additional compounds that have more specificity in targeting the p53 family-dependent metabolic regulation, thus providing anti-cancer therapies with additional tools.
Conclusions
During the past decade, the complex network connecting the p53 family and metabolic pathways has constantly expanded, thereby capturing the attention of the p53/p63/p73 field. Indeed, the tumour suppressive activities of p53, TAp63 and TAp73 do not exclusively rely on their cytostatic and cytotoxic effects, but they are also intertwined with their fundamental roles in governing cellular metabolism (Tomasini et al, 2008; Su et al, 2010, 2012; Li et al, 2012; Rufini et al, 2012). In general, these transcription factors cooperate in supervising glucose and lipid metabolism, mitochondrial functions, as well as ROS production. Many of these activities can also be mimicked by the usage of antidiabetic drugs, such as metformin and pramlintide, whose anticancer efficacy in patients needs to be assessed. Further investigation is also required to determine a possible role of the p53 isoforms in metabolism, as well as to unveil other metabolic pathways potentially regulated by the dominant negative isoforms, ΔNp63 and ΔNp73, and the rest of the family, in addition to what has been reported for glycolysis, synthesis of fatty acids, and ROS production (Sabbisetti et al, 2009; Venkatanarayan et al, 2015). These future characterisations will improve our comprehension of the metabolic regulation by the p53 family as a whole, and may allow the discovery of novel anticancer tools.
References
Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, Huang P (2005) Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO J 24 (19): 3482–3492.
Agarwal ML, Agarwal A, Taylor WR, Chernova O, Sharma Y, Stark GR (1998) A p53-dependent S-phase checkpoint helps to protect cells from DNA damage in response to starvation for pyrimidine nucleotides. Proc Natl Acad Sci USA 95 (25): 14775–14780.
Aldea M, Craciun L, Tomuleasa C, Berindan-Neagoe I, Kacso G, Florian IS, Crivii C (2014) Repositioning metformin in cancer: genetics, drug targets, and new ways of delivery. Tumour Biol 35 (6): 5101–5110.
Ambs S, Ogunfusika MO, Merriam WG, Bennett WP, Billiar TR, Harris CC (1998) Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc Natl Acad Sci USA 95 (15): 8823–8828.
Armata HL, Garlick DS, Sluss HK (2007) The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res 67 (24): 11696–11703.
Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D (2008) SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab 8 (4): 333–341.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126 (1): 107–120.
Bieging KT, Mello SS, Attardi LD (2014) Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14 (5): 359–370.
Boidot R, Vegran F, Meulle A, Le Breton A, Dessy C, Sonveaux P, Lizard-Nacol S, Feron O (2012) Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res 72 (4): 939–948.
Botchkarev VA, Flores ER (2014) p53/p63/p73 in the epidermis in health and disease. Cold Spring Harb Perspect Med 4: 8.
Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rotig A (2007) Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet 39 (6): 776–780.
Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, Karnezis AN, Attardi LD (2011) Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145 (4): 571–583.
Budanov AV, Karin M (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134 (3): 451–460.
Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, Barelier S, Vasseur S, Spoto RP, Pebusque MJ, Dusetti NJ, Iovanna JL, Carrier A (2009) Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res 69 (1): 219–226.
Chakravarti D, Su X, Cho MS, Bui NH, Coarfa C, Venkatanarayan A, Benham AL, Flores Gonzalez RE, Alana J, Xiao W, Leung ML, Vin H, Chan IL, Aquino A, Muller N, Wang H, Cooney AJ, Parker-Thornburg J, Tsai KY, Gunaratne PH, Flores ER (2014) Induced multipotency in adult keratinocytes through down-regulation of DeltaNp63 or DGCR8. Proc Natl Acad Sci USA 111 (5): E572–E581.
Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH (2013) TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell 25 (5): 463–477.
Contractor T, Harris CR (2012) p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res 72 (2): 560–567.
D'Alessandro A, Marrocco C, Rinalducci S, Peschiaroli A, Timperio AM, Bongiorno-Borbone L, Finazzi Agro A, Melino G, Zolla L (2013) Analysis of TAp73-dependent signaling via omics technologies. J Proteome Res 12 (9): 4207–4220.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356 (6366): 215–221.
Duijf PH, Vanmolkot KR, Propping P, Friedl W, Krieger E, McKeon F, Dotsch V, Brunner HG, van Bokhoven H (2002) Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum Mol Genet 11 (7): 799–804.
Flores ER, Lozano G (2012) The p53 family grows old. Genes Dev 26 (18): 1997–2000.
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T (2002) p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416 (6880): 560–564.
Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Borresen-Dale AL, Levine AJ, Bargonetti J, Prives C (2012) Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148 (1-2): 244–258.
Hastak K, Paul RK, Agarwal MK, Thakur VS, Amin AR, Agrawal S, Sramkoski RM, Jacobberger JW, Jackson MW, Stark GR, Agarwal ML (2008) DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc Natl Acad Sci USA 105 (17): 6314–6319.
He Z, Agostini M, Liu H, Melino G, Simon HU (2015) p73 regulates basal and starvation-induced liver metabolism in vivo. Oncotarget 6 (32): 33178–33190.
He Z, Liu H, Agostini M, Yousefi S, Perren A, Tschan MP, Mak TW, Melino G, Simon HU (2013) p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ 20 (10): 1415–1424.
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z (2010) Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA 107 (16): 7455–7460.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4 (1): 1–7.
Jiang P, Du W, Mancuso A, Wellen KE, Yang X (2013a) Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493 (7434): 689–693.
Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X (2011) p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 13 (3): 310–316.
Jiang P, Du W, Yang X (2013b) A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle 12 (24): 3720–3726.
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493 (7432): 338–345.
Joruiz SM, Bourdon JC (2016) p53 isoforms: key regulators of the cell fate decision. Cold Spring Harb Perspect Med 6 (8): a026039.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L (2013) Mutational landscape and significance across 12 major cancer types. Nature 502 (7471): 333–339.
Kawauchi K, Araki K, Tobiume K, Tanaka N (2008) p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol 10 (5): 611–618.
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD (2013) Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27 (9): 1016–1031.
Kim HR, Roe JS, Lee JE, Cho EJ, Youn HD (2013) p53 regulates glucose metabolism by miR-34a. Biochem Biophys Res Commun 437 (2): 225–231.
Kitamura N, Nakamura Y, Miyamoto Y, Miyamoto T, Kabu K, Yoshida M, Futamura M, Ichinose S, Arakawa H (2011) Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS One 6 (1): e16060.
Laezza C, D'Alessandro A, Di Croce L, Picardi P, Ciaglia E, Pisanti S, Malfitano AM, Comegna M, Faraonio R, Gazzerro P, Bifulco M (2015) p53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis 6: e1909.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119 (6): 861–872.
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W (2012) Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149 (6): 1269–1283.
Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM (1996) A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev 10 (8): 934–947.
Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493 (7433): 542–546.
Marchenko ND, Zaika A, Moll UM (2000) Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 275 (21): 16202–16212.
Martinez-Lopez N, Athonvarangkul D, Singh R (2015) Autophagy and aging. Adv Exp Med Biol 847: 73–87.
Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M (2007) Delayed ageing through damage protection by the Arf/p53 pathway. Nature 448 (7151): 375–379.
Mathupala SP, Heese C, Pedersen PL (1997) Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem 272 (36): 22776–22780.
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM (2006) p53 regulates mitochondrial respiration. Science 312 (5780): 1650–1653.
Matusewicz L, Meissner J, Toporkiewicz M, Sikorski AF (2015) The effect of statins on cancer cells—review. Tumour Biol 36 (7): 4889–4904.
Napoli M, Flores ER (2013) The family that eats together stays together: new p53 family transcriptional targets in autophagy. Genes Dev 27 (9): 971–974.
Napoli M, Flores ER (2016) Unifying the p73 knockout phenotypes: TAp73 orchestrates multiciliogenesis. Genes Dev 30 (11): 1253–1254.
Napoli M, Venkatanarayan A, Raulji P, Meyers BA, Norton W, Mangala LS, Sood AK, Rodriguez-Aguayo C, Lopez-Berestein G, Vin H, Duvic M, Tetzlaff MB, Curry JL, Rook AH, Abbas HA, Coarfa C, Gunaratne PH, Tsai KY, Flores ER (2016) DeltaNp63/DGCR8-dependent microRNAs mediate therapeutic efficacy of HDAC inhibitors in cancer. Cancer Cell 29 (6): 874–888.
Okamura S, Ng CC, Koyama K, Takei Y, Arakawa H, Monden M, Nakamura Y (1999) Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol Res 11 (6): 281–285.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T (2004) Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119 (6): 847–860.
Orzol P, Holcakova J, Nekulova M, Nenutil R, Vojtesek B, Coates PJ (2015) The diverse oncogenic and tumour suppressor roles of p63 and p73 in cancer: a review by cancer site. Histol Histopathol 30 (5): 503–521.
Park JH, Zhuang J, Li J, Hwang PM (2016) p53 as guardian of the mitochondrial genome. FEBS Lett 590 (7): 924–934.
Ramis MR, Esteban S, Miralles A, Tan DX, Reiter RJ (2015) Caloric restriction, resveratrol and melatonin: Role of SIRT1 and implications for aging and related-diseases. Mech Ageing Dev 146-148: 28–41.
Restelli M, Lopardo T, Lo Iacono N, Garaffo G, Conte D, Rustighi A, Napoli M, Del Sal G, Perez-Morga D, Costanzo A, Merlo GR, Guerrini L (2014) DLX5, FGF8 and the Pin1 isomerase control DeltaNp63alpha protein stability during limb development: a regulatory loop at the basis of the SHFM and EEC congenital malformations. Hum Mol Genet 23 (14): 3830–3842.
Ruetenik A, Barrientos A (2015) Dietary restriction, mitochondrial function and aging: from yeast to humans. Biochim Biophys Acta 1847 (11): 1434–1447.
Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, Federici M, Dinsdale D, Knight RA, Melino G, Mak TW (2012) TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev 26 (18): 2009–2014.
Sabbisetti V, Di Napoli A, Seeley A, Amato AM, O'Regan E, Ghebremichael M, Loda M, Signoretti S (2009) p63 promotes cell survival through fatty acid synthase. PLoS One 4 (6): e5877.
Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA (2011) Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470 (7334): 359–365.
Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, Tsuchihara K, Bungard D, Berger SL, Jones RG, Mak TW, Zaugg K (2013) Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ 20 (4): 659–668.
Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E (2004) The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 64 (7): 2627–2633.
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, Wistuba I, Flores ER (2010) TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467 (7318): 986–990.
Su X, Chakravarti D, Flores ER (2013) p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer 13 (2): 136–143.
Su X, Gi YJ, Chakravarti D, Chan IL, Zhang A, Xia X, Tsai KY, Flores ER (2012) TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab 16 (4): 511–525.
Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, Biernaskie JA, Sinha S, Prives C, Pevny LH, Miller FD, Flores ER (2009) TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 5 (1): 64–75.
Subbaramaiah K, Altorki N, Chung WJ, Mestre JR, Sampat A, Dannenberg AJ (1999) Inhibition of cyclooxygenase-2 gene expression by p53. J Biol Chem 274 (16): 10911–10915.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, Sugano S, Sato E, Nagao T, Yokote K, Tatsuno I, Prives C (2010) Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 107 (16): 7461–7466.
Timofeev O, Schlereth K, Wanzel M, Braun A, Nieswandt B, Pagenstecher A, Rosenwald A, Elsasser HP, Stiewe T (2013) p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep 3 (5): 1512–1525.
Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A, Tsao MS, Iovanna JL, Squire J, Jurisica I, Kaplan D, Melino G, Jurisicova A, Mak TW (2008) TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 22 (19): 2677–2691.
Tucci P (2012) Caloric restriction: is mammalian life extension linked to p53? Aging (Albany NY) 4 (8): 525–534.
Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Park SH, Thompson T, Karsenty G, Bradley A, Donehower LA (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature 415 (6867): 45–53.
Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A (2013) p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3 (5): 1339–1345.
Venkatanarayan A, Raulji P, Norton W, Chakravarti D, Coarfa C, Su X, Sandur SK, Ramirez MS, Lee J, Kingsley CV, Sananikone EF, Rajapakshe K, Naff K, Parker-Thornburg J, Bankson JA, Tsai KY, Gunaratne PH, Flores ER (2015) IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 517 (7536): 626–630.
Venkatanarayan A, Raulji P, Norton W, Flores ER (2016) Novel therapeutic interventions for p53-altered tumors through manipulation of its family members, p63 and p73. Cell Cycle 15 (2): 164–171.
Vigneron A, Vousden KH (2010) p53, ROS and senescence in the control of aging. Aging (Albany NY) 2 (8): 471–474.
Walerych D, Napoli M, Collavin L, Del Sal G (2012) The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 33 (11): 2007–2017.
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W (2016) Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep 17 (2): 366–373.
Wang X, Zhao X, Gao X, Mei Y, Wu M (2013) A new role of p53 in regulating lipid metabolism. J Mol Cell Biol 5 (2): 147–150.
Warburg O (1925) The metabolism of carcinoma cells. Cancer Res 9 (1): 148–163.
Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, Itie-Youten A, Wakeham A, Arsenian-Henriksson M, Melino G, Kaplan DR, Miller FD, Mak TW (2010) Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev 24 (6): 549–560.
Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Gotoda T, Nagai R, Kimura S, Ishibashi S, Osuga J, Yamada N (2003) p53 Activation in adipocytes of obese mice. J Biol Chem 278 (28): 25395–25400.
Yoon KA, Nakamura Y, Arakawa H (2004) Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet 49 (3): 134–140.
Zhao Y, Chaiswing L, Velez JM, Batinic-Haberle I, Colburn NH, Oberley TD, St Clair DK (2005) p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res 65 (9): 3745–3750.
Zhuang J, Ma W, Lago CU, Hwang PM (2012) Metabolic regulation of oxygen and redox homeostasis by p53: lessons from evolutionary biology? Free Radic Biol Med 53 (6): 1279–1285.
Acknowledgements
ERF is an NCI Outstanding Investigator (R35CA197452), Moffitt Distinguished Scholar, scholar of the Leukemia and Lymphoma Society, the Rita Allen Foundation and the V Foundation for Cancer Research. MN is a CPRIT-TRIUMPH Scholar and was supported by a Research Training Award (RP140106) and Grant (RP150094) from the Cancer Prevention and Research Institute of Texas. We thank DM Aten and JT Pietz for art production.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Napoli, M., Flores, E. The p53 family orchestrates the regulation of metabolism: physiological regulation and implications for cancer therapy. Br J Cancer 116, 149–155 (2017). https://doi.org/10.1038/bjc.2016.384
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.384
Keywords
This article is cited by
-
Does the miR-105–1-Kisspeptin Axis Promote Ovarian Cell Functions?
Reproductive Sciences (2024)
-
Ameliorative effects of bilirubin on cell culture model of non-alcoholic fatty liver disease
Molecular Biology Reports (2023)
-
Role of MicroRNAs, Aptamers in Neuroinflammation and Neurodegenerative Disorders
Cellular and Molecular Neurobiology (2022)
-
The p53 family member p73 in the regulation of cell stress response
Biology Direct (2021)
-
Transcription factor p73 regulates Th1 differentiation
Nature Communications (2020)
