
Abstract
Cancer is a complex disease and progresses within a dynamically evolving extracellular matrix that controls virtually every aspect of the tumour and tumour-associated cells. Interactions with the extracellular microenvironment are predominately mediated by a family of cell-surface transmembrane receptors called integrins. Integrin–matrix engagement leads to the formation of adhesion plaques, consisting of signalling and adaptor proteins, at the plasma membrane that link the extracellular matrix to the regulation of the cell cytoskeleton. In this review, we will highlight exciting data that identify new roles for integrins and integrin-dependent signalling in cancer away from the plasma membrane, discuss the implications of integrin-dependent regulation of Met and ErbB2 growth factor receptors and highlight the role of specific integrins in different stages of cancer development including maintenance of cancer stem cells.
Similar content being viewed by others

Main
Cell adhesion to the extracellular matrix (ECM) is vital for multicellular life and is predominantly mediated by a family of transmembrane cell-surface receptors named integrins. To date, 24 distinct integrin heterodimers, generated from a combination of 18 α- and 8 β-subunits, have been identified in mammals. The specific pattern of integrin expression determines which ECM components a cell can bind to and therefore how a cell senses and responds to its microenvironment.
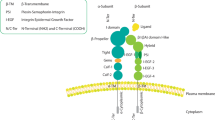
Integrins are unique among transmembrane receptors in that they signal bidirectionally. Recruitment of talin, an adaptor protein, to the integrin tail induces a conformational switch in the integrin extracellular domain, leading to receptor activation and increased affinity for ECM ligands (inside-out signalling). Integrin–ECM engagement, in turn, leads to the recruitment of adaptor and signalling proteins to the integrin cytoplasmic domain and promotes the assembly of macromolecular complexes termed focal adhesions (outside-in signalling) (Figure 1). Through this bidirectional linkage, integrins provide spatially restricted communication lines between the cell and their microenvironment, act as key mechanosensing and force-transducing units and coordinate actin cytoskeletal polymerisation to regulate important biological functions including proliferation, gene expression, cell survival and cell motility (Legate et al, 2009). In the setting of cancer, these same integrin-mediated processes are exploited by tumour cells to promote invasiveness and oncogenic survival and to engineer a host microenvironment that is conducive to tumour growth and metastatic spread. Furthermore, the intricate interplay between integrins, growth factors (GFs) and GF receptors (GFRs) adds an additional level of complexity to ECM-initiated signals and ultimately the control of cell fate (Figure 1) (Ivaska and Heino, 2011) and is often a confounding factor fostering drug resistance in cancer therapies.

Integrins are bidirectional cell-surface signalling molecules. Multiple signals within the cell (signal A) can trigger integrin activation by promoting the recruitment of activators such as talin to the β-integrin cytoplasmic tail. Talin binding to integrins promotes a conformational transition from a bent inactive receptor to an open and primed integrin that favours ligand binding (inside-out signalling). Integrin–ECM interaction, in turn, elicits intracellular signals (signal B), induces focal adhesion formation and actin cytoskeletal reorganisation that affect downstream cellular processes (outside-in signalling). Growth factor receptors are capable of eliciting specific signals (signal C) or in cross-talk with integrins feed into the same pathways to generate unique signals (signal D) in response to ECM cues (outside-in signalling).
Accordingly, disrupting integrin function to treat cancer is an area of extensive and continuing research and several synthetic peptides and humanised antibodies have been developed to interfere with integrin–ligand interactions. Regardless of the encouraging in vitro and preclinical results, late phase clinical trials have, thus far, been disappointing (Desgrosellier and Cheresh, 2010). The recent POSEIDON trial (NCT01008475) showed no improvement in treating patients with K-RAS wild-type metastatic colorectal cancer with abituzumab (anti-αv-integrin antibody) combined with the standard of care (Elez et al, 2015). Similarly, a phase 3 trial with the integrin antagonist cilengitide in combination with radiotherapy did not show any improvement in glioblastoma patient outcome (NCT00689221) and further development of the drug for treating cancer was discontinued (Stupp et al, 2014). Given the complexity of integrin function and integrin-GFR cross-talk, it is possible that targeting signalling molecules downstream of integrins, rather than integrins themselves, would be a more beneficial therapeutic approach. Focal adhesion kinase (FAK), a non-receptor tyrosine kinase implicated in β-integrin signalling, is enriched in focal adhesions and together with Src kinase coordinates adhesion turnover, actin cytoskeleton dynamics and cell shape and regulates cancer cell migration and cell invasion. As such, several clinical trials are currently ongoing with FAK (GSK2256098, PF04554878, VS-4718 and so on) and Src (dasatinib) inhibitors. Currently, the majority of these trials are under phase 1–2 safety and pharmacokinetic evaluation and the true potential of the corresponding drugs will become apparent in the future.
Integrins and long-range signalling in cancer
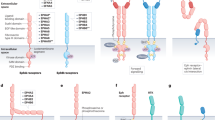
In the classical perception of integrin signalling, integrin activity and function are spatially restricted to the plasma membrane and require an intact uninterrupted integrin–ECM connection. Interestingly, emerging data continue to uncover new and unanticipated roles for core adhesion proteins in cancer-relevant pathways distal to cell–ECM contacts and this is exemplified by the core adhesome kinase FAK (Figure 2). FAK, a recognised focal adhesion-localised protein that is activated in response to integrin–ECM engagement, has also been detected in active-integrin-positive early endosomes. FAK was found to be active in endosomes and to prolong integrin–ECM-initiated signals following receptor internalisation to suppress anoikis and support anchorage-independent growth of breast cancer cells in vitro and to promote metastasis in vivo (Figure 2A) (Alanko et al, 2015). These data are intriguing on two fronts. First, they challenge the classical view of integrin signalling, thus opening up new avenues to target integrin function where defining the nature (i.e. composition and dynamics) of active–integrin–endosomal complexes will be fundamental and, second, suggest that cancer cells may be able to highjack and repurpose integrin trafficking pathways to prolong ECM-dependent survival cues in the absence of a physical cell–ECM contact. This may be one mechanism employed by circulating tumour cells to avoid anoikis initiation during metastatic spread. FAK and Src, in addition to talin, have been observed in other endosomal compartments containing unligated active integrins and found to be important for coordinating directional cell migration in fibroblasts by maintaining integrin activity and focal adhesion polarity (Nader et al, 2016) (Figure 2A). Although the relevance of this endocytic pathway was not investigated in the context of cancer, regulation of integrin activity in endosomes may offer an ideal opportunity for cancer cells to increase migration efficiency during cell invasion.

Integrins and integrin-related signalling contribute to cancer progression from different organelles. (A) Integrin signalling is maintained in endosomes away from the plasma membrane and contributes to anoikis resistance in cancer (left panel) and to focal adhesion polarisation and directional cell migration in fibroblasts (right panel). Key endosomal and adhesion-related proteins that contribute to these processes are indicated. Rab21 is a marker of early endosomes and Rab11 denotes perinuclear recycling compartments. (B) Nuclear localisation of active FAK (left panel) promotes transcription of immunomodulatory genes such as Ccl5 and tgfb that allow tumour cells to evade the immune system. ZNF304-mediated transcriptional upregulation of ITGB1 (right panel) promotes downstream integrin signalling and contributes to anoikis resistance in cancer. (C) At the plasma membrane, Cyclin-D1/Cdk4-mediated phosphorylation of paxillin triggers Rac activity, membrane ruffling and cell invasion. Focal adhesions and focal complex localisation and actin stress fibres are illustrated in membrane ruffles in the top inset. (D) Tumour cell secretion and intercellular transfer of αvβ6-containing exosomes facilitates aggressive disease propagation. (E) Tumour-secreted exosomes containing specific integrin signatures predetermine the site of metastatic dissemination and aid in the formation of a premetastatic niche (left panel). Exosomal release of TGF-β also contributes to the making of a tumour microenvironment (right panel). (F) Filopodia, actin-rich finger-like protrusions, contribute to cancer cell invasion, proliferation and the formation of micrometastases through integrin-dependent adhesion and FAK signalling. Myosin-X, a motor protein that transports integrins to filopodia tips, is associated with cancer cell invasion and poor patient prognosis. An adhesion plaque is indicated at the filopodia tip. Ccl5, chemokine (C–C motif) ligand 5; EEA1, early endosome antigen 1; ITGB1, integrin β1; PIPKγ, type I phosphatidylinositol phosphate kinase; Rab, Ras-related proteins in brain (GTPase signalling molecules); tgfb, transforming growth factor β.
Focal adhesion kinase has also been detected in cell nuclei of squamous cell carcinoma in association with chromatin and other transcription factors. This cancer cell-specific nuclear-localised FAK was shown to act as a scaffold to regulate another cancer hallmark, that is, the transcription of immunomodulatory genes in favour of tumour tolerance in a process requiring FAK kinase activity (Serrels et al, 2015) (Figure 2B). Targeting FAK signalling in pancreatic cancer was recently demonstrated to increase immune cell infiltration into the tumour microenvironment and to sensitise pancreatic tumours to checkpoint immunotherapy (Jiang et al, 2016). Although in this work FAK localisation was not investigated, together these studies highlight intriguing examples of potential integrin-immune system cross-talk in cancer and the value of targeting FAK in combination with immunotherapy. Understanding the specific stimuli or signalling pathways that drive FAK to the nucleus in different cancers will be vital in developing an effective antitumour approach. Current hypotheses propose cellular stress, disengagement of integrin–ECM contacts and kinase inhibition as contenders that increase levels of ‘free’ FAK and FAK nuclear accumulation (Lim, 2013).
It is important to note that, in addition to FAK, other canonical integrin adhesome scaffolding proteins including α-actinin, paxillin, zyxin and related family members have been observed in cell nuclei and several have been implicated in gene transcription (Hervy et al, 2006; Byron and Frame, 2016). Whether the nuclear roles of these proteins are linked to their function in adhesion plaques and the implications for disease (e.g. cancer) remain to be determined. These studies, however, emphasise the need to further understand if or how subcellular compartmentalisation regulates the function of integrin adhesome proteins in pathophysiological settings.
In ovarian cancer, transcriptional upregulation of β1-integrin by ZNF304 (a zinc-finger transcription factor) and subsequent activation of a Src–FAK–paxillin axis was demonstrated to enforce anoikis resistance in vitro and ZNF304 gene silencing to significantly inhibit ovarian tumour growth in a mouse model (Aslan et al, 2015) (Figure 2B). It would be interesting to determine if endosomal accumulation of active integrins and/or nuclear localisation of FAK or paxillin contribute to the observed ZNF304-mediated effects. Nevertheless, the enhanced antitumour efficacy of ZNF304 silencing in combination with paclitaxel suggests that targeting integrin transcription may provide complementary treatment for ovarian cancer (Aslan et al, 2015).
In addition to new distal roles for focal adhesion proteins in protumourigenic processes, known cancer-associated proteins, have been shown to achieve prodisease effects through modulation of pathways intimately linked with cell adhesion. For example, cyclin D1, a prominent transcriptional cell cycle regulator and instigator of transformation and tumorigenesis, has recently been shown to bind to filamin A, Rho and PACSIN2 (components of the integrin adhesome) in the cytoplasm and in complex with its kinase binding partner Cdk4 (another integrin adhesome protein) to phosphorylate a subset of membrane-associated paxillin in membrane ruffles. Phosphorylation of paxillin, in turn, triggers Rac GTPase activity (cytoskeletal regulator), membrane ruffling and cell invasion in normal fibroblasts and tumour cells in vitro (Fuste et al, 2016) (Figure 2C). Intriguingly, the pool of cyclin-D1/Cdk4-phosphorylated paxillin was notably absent from focal adhesions; however, the possibility that it was localised in nascent integrin adhesions (focal complexes) in membrane ruffles was not determined.
More extreme examples of long-range integrin signalling have been illustrated in tumour exosomes and associated with disease advancement. Intercellular exosome transfer of the αvβ6-integrin heterodimer has been suggested to contribute to cancer progression by facilitating horizontal propagation of more aggressive integrin-associated prostate cancer phenotypes (Fedele et al, 2015) (Figure 2D). Moreover, biodistribution and proteome analyses of tumour-secreted exosomes have revealed specific integrin-heterodimer signatures that direct metastases to different organs by fusing with the target tissue and initiating the formation of a premetastatic niche. In particular, exosomal α6β1- and α6β4-integrins were associated with lung metastasis and αvβ5-integrin was linked to liver metastasis (Hoshino et al, 2015) (Figure 2E). In addition to integrins, transforming growth factor-β1 (TGF-β1) has been located in tumour-derived exosomes where it promotes transformation of bone marrow mesenchymal stem cells to tumour-supportive myofibroblasts (Chowdhury et al, 2015) (Figure 2E). As αv-integrins are known regulators of TGF-β activity, it is possible that exosomal integrins and TGF-β cooperate to prepare the metastatic niche. These highly interesting discoveries suggest a previously unappreciated role for integrins in cancer and shed light on Paget’s seed-to-soil hypothesis offering the potential for novel prognostic and therapeutic interventions.
Integrins and the tumour microenvironment
As bidirectional signalling molecules, integrins are ideally situated to sense, modulate and respond to changes in extracellular stimuli. Cancer cells migrating through the surrounding stroma use distinct integrin-containing actin-rich plasma membrane protrusions such as lamellipodia and motor protein myosin-X-induced filopodia to anchor the cell, probe the microenvironment and promote cell invasion. In particular, β1-integrin-containing filopodium-like protrusions have been implicated in activating dormant tumour cells and promoting proliferation and formation of micrometastases at distal sites (Shibue et al, 2012; Jacquemet et al, 2015) (Figure 2F).
Although normal stroma has recently been shown to impede tumorigenesis (Kaukonen et al, 2016), the biophysical and biochemical cues from a tumour microenvironment support malignancy by modulating the hallmarks of cancer (Bissell and Hines, 2011; Pickup et al, 2014). Cancer-associated stromal fibroblasts have been viewed as the protagonists of a dynamically evolving cancer microenvironment and promoters of tumour growth and progression. Recently, ‘paradoxical’ activation of melanoma-associated fibroblasts by PLX4720, a BRAF inhibitor, was demonstrated to increase stromal matrix production and stiffening. Importantly, these ECM changes triggered elevated β1-integrin-Src-FAK signalling in melanoma tumour cells and contributed to BRAF-mutant melanoma resistance to PLX4720 (Hirata et al, 2015). Notably, in the absence of stromal fibroblasts and on soft matrices, isolated melanoma spheroids/cells remained susceptible to BRAF inhibition, whereas increasing the elastic modulus of the matrix was sufficient to subdue the apoptotic response. Here, coinhibition of BRAF and FAK was found to lead to more effective control of BRAF-mutant melanoma. These data demonstrate the acute impact of the tumour microenvironment on tumour cell signalling and acquisition of resistance.
In recent years, microscopy techniques including two-photon second-harmonic generation have strengthened the correlation between the stiffness and organisation of the tumour stroma and disease progression (Conklin et al, 2011; Laklai et al, 2016; Oudin et al, 2016). In human breast carcinoma, a specific tumour-associated collagen signature (TACS) consisting of straight, aligned collagen fibre bundles, perpendicular to the tumour boundary, was found to correlate with patient survival (Conklin et al, 2011). Similarly, in pancreatic ductal adenocarcinoma, increased periductal collagen thickness, high epithelial tension and matricellular fibrosis were demonstrated to be indicative of an aggressive disease state (Laklai et al, 2016). Importantly, modulation of tumour cell tension, dictated by the tumour genotype, was found to promote matrix remodelling in response to elevated β1-integrin mechanotransduction and loss of TGF-β signalling (Laklai et al, 2016). These data highlight an interesting concept whereby tumour cells harbour an intrinsic ability, encoded within their genome, to differentially tune their matrix mechanics towards prometastasis and potentiation of disease state. Furthermore, it appears that biomechanical signalling downstream of integrins has an important role in translating tumour cell-specific signals in this process.
Integrins have also been implicated in tumour-mediated modification of the microenvironment in breast cancer. Direct interaction between the prometastatic isoform of the actin regulatory protein Mena (MenaINV) and α5β1-integrin enhanced three-dimensional haptotaxis of tumour cells towards a fibronectin gradient in vitro and in vivo and contributed to collagen network remodelling into thin capsules perpendicular to the tumour edge (Oudin et al, 2016). However, the similarity of these collagen capsules to the TACS identified in previous studies and the mechanism whereby α5β1-integrin, a fibronectin-specific receptor, may affect collagen fibrillogenesis was not investigated. Of note, in non-small-cell lung carcinoma, expression of α11β1-integrin (a stromal cell-specific collagen receptor) in cancer-associated fibroblasts was found to correlate with collagen crosslinking and tumour stiffness and to promote tumour metastasis (Navab et al, 2016).
In breast myoepithelial cells, differences in the bond dynamics of fibronectin receptors α5β1- and αvβ6-integrins have been implicated in sensing matrix rigidities associated with healthy and malignant tissues, respectively (Elosegui-Artola et al, 2014). Interestingly, the β6-integrin subunit is selectively expressed in development and wound healing, suggesting an important role for the αvβ6 receptor during tissue remodelling. In addition, αvβ6-integrin is upregulated in a variety of carcinomas and strongly associated with cell migration, invasion and survival, particularly in breast cancer and primarily through the activation of TGF-β. As TGF-β activation is a key instigator of fibrosis and matrix remodelling, perhaps differential binding of αvβ6- over α5β1-integrin to stiffer matrices serves to positively reinforce tumour-specific TGF-β activation by promoting cell-surface localisation of the main TGF-β activator. Notably, blocking αvβ6 function alone or together with trastuzumab has been proposed as an alternative approach to treat high-risk and trastuzumab-resistant breast cancer patients (Moore et al, 2014).
Integrins and GF synergy in cancer progression and acquisition of drug resistance
Integrin signalling is closely coupled to the function and activity of many different GFRs, whether by direct activation of GFs such as TGF-β or by feeding into the same signalling axes as GFRs such as phosphoinositide 3-kinase/AKT and mitogen-activated protein kinase pathways (Ivaska and Heino, 2011). Thus, integrin function can also determine how cells respond to GFs and as such has been implicated in the acquisition of resistance to anti-GFR therapies. In these cases, providing an effective combinatorial treatment that tackles integrin-GFR cross-talk will require in-depth knowledge of the specific integrin subunits involved. For example, although both β1- and β4-integrins have been implicated in the signalling of the same GFRs and in the development of therapeutic resistance, these receptors are structurally distinct and are unlikely to regulate GFRs through similar mechanisms.
For example, β1-integrin has been linked to anti-ErbB2 therapy (trastuzumab and lapatinib) resistance in breast cancer (Lesniak et al, 2009; Huang et al, 2011) and the α3β1-integrin heterodimer, in complex with the tetraspanin CD151, has been demonstrated to promote ErbB2 homodimerization and ErbB2-driven proliferation in laminin-rich ECMs (Novitskaya et al, 2014). On the other hand, integrin β4, a unique β-subunit with a large cytoplasmic domain harbouring kinase activity and lacking the typical binding motifs and actin linkage of other β-integrins, has been shown to form a complex with ErbB2 and in this way enhance STAT3/c-Jun signalling and facilitate ErbB2-driven tumorigenesis in breast cancer (Guo et al, 2006). Here, the deletion of the β4-integrin cytoplasmic domain increased the efficacy of ErbB2-targeted therapy.
Although β1-integrin in a complex with tensin-4 and Met maintains the stability of Met (Muharram et al, 2014), another clinically relevant oncogenic GFR, α6β4-integrin is important for HGF-mediated activation of c-Met to drive tumorigenesis and cancer cell invasion (Trusolino et al, 2001). A β1-integrin–Met complex has also been recently described in endomembranes where it facilitates sustained HGF-induced extracellular signal-regulated kinase activation via ‘inside-in signalling’ (Barrow-McGee et al, 2016).
Interestingly, from a therapeutic point of view, β4-mediated regulation of both Met and ErbB2 appears to occur independently of integrin–ligand binding (Trusolino et al, 2001; Guo et al, 2006). This is also the case for β1-integrin–Met endosomal cosignalling, which is adhesion independent (Barrow-McGee et al, 2016). Accordingly, traditional integrin-targeting therapeutic antibodies or adhesion-blocking peptides will not be effective in these cases. On the contrary, β1-mediated activation of ErbB2 was mediated mainly by the canonical integrin pathway feeding into ErbB2 signalling and this was prevented by targeting β1 with therapeutic antibodies. These few examples highlight the complexity of integrin-mediated regulation of GFR function and in choosing an appropriate therapeutic approach. In one case targeting the ligand binding extracellular domain of integrins may be sufficient to disrupt integrin-GFR cross-talk; however, in other cases, interfering with integrin downstream signalling may be more effective. In the latter approach, detailed understanding of the adaptors and/or signalling proteins recruited to the distinct β1- and β4-integrin subunits will be essential.
Integrins and regulation of cancer stem cell properties during metastasis
Cancer stem cells (CSCs), also known as tumour-initiating cells, are a small therapeutically resistant, self-renewing sub-population of tumour cells responsible for long-term tumour propagation and metastasis (Medema, 2013). Not surprisingly, specific integrins have an important role in the maintenance of stem cell properties in normal tissue and in the context of cancer (Medema, 2013). CSCs are dependent on integrin signalling triggered by specific ECM proteins such as periostin and tenascin C, which are enriched in the so-called stem cell niche. Several integrin subunits including β3, α6 and β1 are important for the self-renewal and maintenance of the CSC phenotype at the primary tumour and additionally serve as CSC markers (Seguin et al, 2015). The role of integrins in CSCs has recently been extensively covered (Seguin et al, 2015) and we will only highlight a few examples here. Integrin β3 has been suggested to be important for stemness and in facilitating resistance to tyrosine kinase inhibitors (Seguin et al, 2015). Integrin α6/β1 is essential for maintenance of CSC sub-populations (Lathia et al, 2010; Goel et al, 2013); it activates BMI-1 (B-cell-specific Moloney murine leukaemia virus integration site 1, a stem cell factor, and a polycomb group family member), via FAK, to support tumour initiation and self-renewal of breast CSCs (Goel et al, 2013). In glioblastoma CSCs, α6 was found to be coexpressed only with β1 (not with β4) and with proximal extracellular laminin. Furthermore, depletion of α6-integrin reduces CSC numbers, indicating a role for this integrin in CSC maintenance in the primary tumour and that targeting α6 could be a promising therapeutic option (Lathia et al, 2010).
Following tumour cell dissemination to a secondary site, metastatic cells need to re-establish a new growth-supportive niche as the new tissue microenvironment is fundamentally distinct from the primary tumour site. The requirement for microenvironment remodelling is most likely linked to the notion that disseminated tumour cells can remain dormant for many years or even decades. Single-cell sequencing of metastatic breast cancer cells has revealed that the first cells to disseminate and form early-stage ‘low-burden’ metastases are enriched for genes characteristic of CSCs, whereas later stage bigger ‘high-burden’ metastases are more similar to the primary tumour (Lawson et al, 2015). Somewhat surprisingly expression of the β1-integrin subunit was suppressed in low-burden CSCs, whereas α6-integrin was upregulated. However, β1-integrin expression may have an important function in the extravasation of metastatic cells (Chen et al, 2016). These data suggest that CSCs may use different integrins to maintain their stem cell properties between the primary and distant metastatic sites, as β1-integrin normally linked to CSCs in the primary tumour is downregulated in low-burden metastatic CSCs. Adhesion to supportive ECM may be essential in the early steps of metastatic colonization as disseminating CSCs remain in contact with endothelial cells for a long period of time after extravasation (Malladi et al, 2016). Previously, endothelial cell periostin and thrombospondin-1 secretion has been shown to regulate the dormancy and proliferation of metastatic breast cancer cells (Ghajar et al, 2013). Accordingly, the interaction between ECM proteins secreted by endothelial cells and the integrin receptors expressed by CSCs may be important for the maintenance of CSC properties at distant sites during the early steps of colonization. However, once metastases start to grow, tumour cells can reform the CSC niche at secondary sites by secreting β1- and β3-integrin ligands, such as tenascin C, which promote self-renewal of CSCs, as has been shown during colonization of breast cancer cells (Oskarsson et al, 2011).
Conclusion
Deregulation of integrin-mediated cell adhesion to the ECM is implicated at every step of tumorigenesis from initial hyperproliferation to local invasion, metastatic dissemination and colonisation of distant organs. The complex cross-talk between the tumour and its microenvironment is increasingly recognised as a key contributor to cancer progression. Initial attempts to target integrins or their downstream signalling in cancer have not been clinically beneficial. However, growing appreciation of the diverse biological roles of integrins in cancer are likely to facilitate design of better targeting agents and to improve stratification of patients most likely to respond to such therapeutics. Here, we have particularly highlighted recent advances in our understanding of unconventional integrin signalling. Integrin-linked signalling away from the plasma membrane in the endosomes, in the nucleus and in mechanosensitive responses to cancer therapy are likely to pave the way for new translational approaches to successfully target integrins in cancer.
References
Alanko J, Mai A, Jacquemet G, Schauer K, Kaukonen R, Saari M, Goud B, Ivaska J (2015) Integrin endosomal signalling suppresses anoikis. Nat Cell Biol 17: 1412–1421.
Aslan B, Monroig P, Hsu MC, Pena GA, Rodriguez-Aguayo C, Gonzalez-Villasana V, Rupaimoole R, Nagaraja AS, Mangala S, Han HD, Yuca E, Wu SY, Ivan C, Moss TJ, Ram PT, Wang H, Gol-Chambers A, Ozkayar O, Kanlikilicer P, Fuentes-Mattei E, Kahraman N, Pradeep S, Ozpolat B, Tucker S, Hung MC, Baggerly K, Bartholomeusz G, Calin G, Sood AK, Lopez-Berestein G (2015) The ZNF304–integrin axis protects against anoikis in cancer. Nat Commun 6: 7351.
Barrow-McGee R, Kishi N, Joffre C, Menard L, Hervieu A, Bakhouche BA, Noval AJ, Mai A, Guzman C, Robert-Masson L, Iturrioz X, Hulit J, Brennan CH, Hart IR, Parker PJ, Ivaska J, Kermorgant S (2016) Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat Commun 7: 11942.
Bissell MJ, Hines WC (2011) Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17: 320–329.
Byron A, Frame MC (2016) Adhesion protein networks reveal functions proximal and distal to cell-matrix contacts. Curr Opin Cell Biol 39: 93–100.
Chen MB, Lamar JM, Li R, Hynes RO, Kamm RD (2016) Elucidation of the roles of tumor integrin beta1 in the extravasation stage of the metastasis cascade. Cancer Res 76: 2513–2524.
Chowdhury R, Webber JP, Gurney M, Mason MD, Tabi Z, Clayton A (2015) Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 6: 715–731.
Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ (2011) Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol 178: 1221–1232.
Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10: 9–22.
Elez E, Kocakova I, Hohler T, Martens UM, Bokemeyer C, Van Cutsem E, Melichar B, Smakal M, Csoszi T, Topuzov E, Orlova R, Tjulandin S, Rivera F, Straub J, Bruns R, Quaratino S, Tabernero J (2015) Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: the randomised phase I/II POSEIDON trial. Ann Oncol 26: 132–140.
Elosegui-Artola A, Bazellieres E, Allen MD, Andreu I, Oria R, Sunyer R, Gomm JJ, Marshall JF, Jones JL, Trepat X, Roca-Cusachs P (2014) Rigidity sensing and adaptation through regulation of integrin types. Nat Mater 13: 631–637.
Fedele C, Singh A, Zerlanko BJ, Iozzo RV, Languino LR (2015) The alphavbeta6 integrin is transferred intercellularly via exosomes. J Biol Chem 290: 4545–4551.
Fuste NP, Fernandez-Hernandez R, Cemeli T, Mirantes C, Pedraza N, Rafel M, Torres-Rosell J, Colomina N, Ferrezuelo F, Dolcet X, Gari E (2016) Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat Commun 7: 11581.
Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, Chen EI, Lyden D, Bissell MJ (2013) The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15: 807–817.
Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, Kumar P, Vander Kooi CW, Shultz LD, Greiner DL, Norum JH, Toftgard R, Kuperwasser C, Mercurio AM (2013) GLI1 regulates a novel neuropilin-2/alpha6beta1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol Med 5: 488–508.
Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, Giancotti FG (2006) Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126: 489–502.
Hervy M, Hoffman L, Beckerle MC (2006) From the membrane to the nucleus and back again: bifunctional focal adhesion proteins. Curr Opin Cell Biol 18: 524–532.
Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, Sahai E (2015) Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27: 574–588.
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527: 329–335.
Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, Shou J, Bissell MJ, Osborne CK, Schiff R (2011) beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res 13: R84.
Ivaska J, Heino J (2011) Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol 27: 291–320.
Jacquemet G, Hamidi H, Ivaska J (2015) Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol 36: 23–31.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG (2016) Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22: 851–860.
Kaukonen R, Mai A, Georgiadou M, Saari M, De Franceschi N, Betz T, Sihto H, Ventela S, Elo L, Jokitalo E, Westermarck J, Kellokumpu-Lehtinen PL, Joensuu H, Grenman R, Ivaska J (2016) Normal stroma suppresses cancer cell proliferation via mechanosensitive regulation of JMJD1a-mediated transcription. Nat Commun 7: 12237.
Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM (2016) Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med 22: 497–505.
Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, Rich JN (2010) Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 6: 421–432.
Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z (2015) Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526: 131–135.
Legate KR, Wickstrom SA, Fassler R (2009) Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev 23: 397–418.
Lesniak D, Xu Y, Deschenes J, Lai R, Thoms J, Murray D, Gosh S, Mackey JR, Sabri S, Abdulkarim B (2009) Beta1-integrin circumvents the antiproliferative effects of trastuzumab in human epidermal growth factor receptor-2-positive breast cancer. Cancer Res 69: 8620–8628.
Lim ST (2013) Nuclear FAK: a new mode of gene regulation from cellular adhesions. Mol Cells 36: 1–6.
Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massague J (2016) Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165: 45–60.
Medema JP (2013) Cancer stem cells: the challenges ahead. Nat Cell Biol 15: 338–344.
Moore KM, Thomas GJ, Duffy SW, Warwick J, Gabe R, Chou P, Ellis IO, Green AR, Haider S, Brouilette K, Saha A, Vallath S, Bowen R, Chelala C, Eccles D, Tapper WJ, Thompson AM, Quinlan P, Jordan L, Gillett C, Brentnall A, Violette S, Weinreb PH, Kendrew J, Barry ST, Hart IR, Jones JL, Marshall JF (2014) Therapeutic targeting of integrin alphavbeta6 in breast cancer. J Natl Cancer Inst 106: dju169.
Muharram G, Sahgal P, Korpela T, De Franceschi N, Kaukonen R, Clark K, Tulasne D, Carpen O, Ivaska J (2014) Tensin-4-dependent MET stabilization is essential for survival and proliferation in carcinoma cells. Dev Cell 29: 421–436.
Nader GP, Ezratty EJ, Gundersen GG (2016) FAK, talin and PIPKIgamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat Cell Biol 18: 491–503.
Navab R, Strumpf D, To C, Pasko E, Kim KS, Park CJ, Hai J, Liu J, Jonkman J, Barczyk M, Bandarchi B, Wang YH, Venkat K, Ibrahimov E, Pham NA, Ng C, Radulovich N, Zhu CQ, Pintilie M, Wang D, Lu A, Jurisica I, Walker GC, Gullberg D, Tsao MS (2016) Integrin alpha11beta1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 35: 1899–1908.
Novitskaya V, Romanska H, Kordek R, Potemski P, Kusinska R, Parsons M, Odintsova E, Berditchevski F (2014) Integrin alpha3beta1-CD151 complex regulates dimerization of ErbB2 via RhoA. Oncogene 33: 2779–2789.
Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massague J (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17: 867–874.
Oudin MJ, Jonas O, Kosciuk T, Broye LC, Guido BC, Wyckoff J, Riquelme D, Lamar JM, Asokan SB, Whittaker C, Ma D, Langer R, Cima MJ, Wisinski KB, Hynes RO, Lauffenburger DA, Keely PJ, Bear JE, Gertler FB (2016) Tumor cell-driven extracellular matrix remodeling drives haptotaxis during metastatic progression. Cancer Discov 6: 516–531.
Pickup MW, Mouw JK, Weaver VM (2014) The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 15: 1243–1253.
Seguin L, Desgrosellier JS, Weis SM, Cheresh DA (2015) Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol 25: 234–240.
Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, Gomez-Cuadrado L, Canel M, Muir M, Ring JE, Maniati E, Sims AH, Pachter JA, Brunton VG, Gilbert N, Anderton SM, Nibbs RJ, Frame MC (2015) Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 163: 160–173.
Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA (2012) The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov 2: 706–721.
Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, Steinbach JP, Wick W, Tarnawski R, Nam DH, Hau P, Weyerbrock A, Taphoorn MJ, Shen CC, Rao N, Thurzo L, Herrlinger U, Gupta T, Kortmann RD, Adamska K, McBain C, Brandes AA, Tonn JC, Schnell O, Wiegel T, Kim CY, Nabors LB, Reardon DA, van den Bent MJ, Hicking C, Markivskyy A, Picard M, Weller M European Organisation for Research and Treatment of Cancer (EORTC), Canadian Brain Tumor Consortium, CENTRIC study team (2014) Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 15: 1100–1108.
Trusolino L, Bertotti A, Comoglio PM (2001) A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 107: 643–654.
Acknowledgements
We apologise to all colleagues whose work could not be cited here due to space constraints. Work in the author’s laboratory was supported by funding from the Academy of Finland, an ERC Consolidator Grant, the Sigrid Juselius Foundation and the Cancer Society of Finland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hamidi, H., Pietilä, M. & Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br J Cancer 115, 1017–1023 (2016). https://doi.org/10.1038/bjc.2016.312
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.312
Keywords
This article is cited by
-
Induction of promyelocytic leukemia zinc finger protein by miR-200c-3p restores sensitivity to anti-androgen therapy in androgen-refractory prostate cancer and inhibits the cancer progression via down-regulation of integrin α3β4
Cellular Oncology (2023)
-
Alternative splicing associated with cancer stemness in kidney renal clear cell carcinoma
BMC Cancer (2021)
-
Leukaemia: a model metastatic disease
Nature Reviews Cancer (2021)
-
DFF40-iRGD, a novel chimeric protein with efficient cytotoxic and apoptotic effects against triple-negative breast cancer cells
Biotechnology Letters (2021)
-
Conditional knockdown of integrin beta-3 reveals its involvement in osteolytic and soft tissue lesions of breast cancer skeletal metastasis
Journal of Cancer Research and Clinical Oncology (2021)
