
Abstract
Background:
Our recent studies of microRNA (miRNA) expression signatures demonstrated that microRNA-29s (miR-29s; miR-29a/b/c) were significantly downregulated in head and neck squamous cell carcinoma (HNSCC) and were putative tumour-suppressive miRNAs in human cancers. Our aim in this study was to investigate the functional significance of miR-29s in cancer cells and to identify novel miR-29s-mediated cancer pathways and responsible genes in HNSCC oncogenesis and metastasis.
Methods:
Gain-of-function studies using mature miR-29s were performed to investigate cell proliferation, migration and invasion in two HNSCC cell lines (SAS and FaDu). To identify miR-29s-mediated molecular pathways and targets, we utilised gene expression analysis and in silico database analysis. Loss-of-function assays were performed to investigate the functional significance of miR-29s target genes.
Results:
Restoration of miR-29s in SAS and FaDu cell lines revealed significant inhibition of cancer cell migration and invasion. Gene expression data and in silico analysis demonstrated that miR-29s modulated the focal adhesion pathway. Moreover, laminin γ2 (LAMC2) and α6 integrin (ITGA6) genes were candidate targets of the regulation of miR-29s. Luciferase reporter assays showed that miR-29s directly regulated LAMC2 and ITGA6. Silencing of LAMC2 and ITGA6 genes significantly inhibited cell migration and invasion in cancer cells.
Conclusion:
Downregulation of miR-29s was a frequent event in HNSCC. The miR-29s acted as tumour suppressors and directly targeted laminin–integrin signalling. Recognition of tumour-suppressive miRNA-mediated cancer pathways provides new insights into the potential mechanisms of HNSCC oncogenesis and metastasis and suggests novel therapeutic strategies for the disease.
Similar content being viewed by others

Main
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world and ∼500 000 cases are diagnosed every year (Jemal et al, 2010). In spite of considerable advances in multimodality therapy, including surgery, radiotherapy and chemotherapy, the overall 5-year survival rate for patients with HNSCC is only 40–50% (Leemans et al, 2011). Local tumour recurrence and distant metastasis after conventional therapy appear to be major contributing factors for restricted survival of HNSCC patients. Therefore, understanding the molecular pathways of metastasis accompanying HNSCC would help to improve diagnosis, approaches to therapy and prevention of the disease.
The discovery of noncoding RNAs in the human genome was an important conceptual breakthrough in the postgenome sequencing era (Mattick, 2004). The microRNAs (miRNAs) constitute a class of small, noncoding RNA molecules (19–22 nucleotides in length) that regulate protein-coding gene expression by repressing translation or cleaving RNA transcripts in a sequence-specific manner (Bartel, 2004). The miRNAs are unique in their ability to regulate multiple protein-coding genes. Bioinformatic predictions indicate that miRNAs regulate >30% of the protein-coding genes in the human genome (Lewis et al, 2005).
A growing body of evidence suggests that miRNAs contribute significantly to HNSCC progression, development and metastasis (Esquela-Kerscher and Slack, 2006). In cancer pathways, normal regulatory mechanisms are disrupted by altered expression of tumour-suppressive or oncogenic miRNAs. Therefore, identification of differentially expressed miRNAs is an important step to understand human oncogenesis. Genome-wide miRNA expression signatures have been used to rapidly and precisely identify aberrant miRNA expression in the HNSCC. Based on recent reports, including our own signatures, several down- or upregulated miRNAs have been discovered in the HNSCC (Avissar et al, 2009; Hui et al, 2010; Kikkawa et al, 2010; Nohata et al, 2011).
Our expression signature of HNSCC demonstrated that the microRNA-29 family (miR-29s) was downregulated in cancer tissues (Kikkawa et al, 2010; Nohata et al, 2011). The miR-29s include three members, miR-29a, miR-29b and miR-29c, in the human genome. The miR-29b includes two members, miR-29b-1 and miR-29b-2 (Wang et al, 2013). These miRNAs form clusters on two human genome loci: miR-29b-1 and miR-29a in 7q32 and miR-29b-2 and miR-29c in 1q32 (Kriegel et al, 2012). Although aberrant expression of miR-29s was reported in several types of human cancers, the expression status of miR-29s varied according to the cancer types (Fabbri et al, 2007; Sengupta et al, 2008; Gebeshuber et al, 2009; Han et al, 2010; Mott et al, 2010; Xiong et al, 2010; Zhao et al, 2010; Li et al, 2011; Fu et al, 2012; Presneau et al, 2013). Therefore, the aim of this study was to investigate the functional significance of the miR-29s in cancer cells and to identify novel miR-29s-regulated cancer pathways and responsible genes in HNSCC oncogenesis and metastasis.
Genome-wide gene expression analysis of miR-29a transfectants and in silico database analysis showed that the focal adhesion pathway was a promising candidate target pathway. In this regard, the laminin–integrin signalling is critically important. It occurs in the basal lamina and influences cell differentiation, migration and adhesion as well as proliferation and cell survival. Interestingly, laminin γ2 (coded by LAMC2), a member of laminin-332, and α6 integrin (coded by ITGA6) were listed as candidate genes in the focal adhesion pathway. Moreover, an interaction between laminin-332 and α6β4 integrin triggers a number of signalling cascades, promoting both cell migration and cancer cell survival (Marinkovich, 2007; Kariya et al, 2009). We investigated the functional significance of LAMC2 and ITGA6 in HNSCC and showed that these genes were regulated by miR-29s. The identification of novel cancer pathways regulated by tumour-suppressive miR-29s provides new insights into the potential mechanisms of HNSCC oncogenesis and metastasis.
Materials and methods
Clinical HNSCC specimens
A total of 23 pairs of primary HNSCC and corresponding normal epithelial samples were obtained from patients with HNSCC at Chiba University Hospital (Chiba, Japan) from 2005 to 2011. The samples considered normal were free of cancer cells by pathologic examination. The patients’ backgrounds and clinicopathological characteristics are summarised in Supplementary Table 1. The patients were classified according to the 2002 Union for International Cancer Control (UICC) TNM staging criteria before treatment. Written consent for tissue donation for research purposes was obtained from each patient before tissue collection. The protocol was approved by the institutional review board of the Chiba University. The specimens were immersed in RNAlater (Qiagen, Valencia, CA, USA) and stored at −20 °C until RNA was extracted.
RNA isolation
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The RNA concentrations were determined spectrophotometrically, and molecular integrity was checked by gel electrophoresis. The quality of RNA was confirmed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
HNSCC cell culture
The following human HNSCC cell lines were used: SAS (derived from a primary lesion of tongue SCC) and FaDu (derived from a primary lesion of hypopharyngeal SCC). Both cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37 °C.
Quantitative real-time RT–PCR (qRT–PCR)
First-strand cDNA was synthesised from 1 μg of total RNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Gene-specific PCR products were assayed continuously using a 7900-HT Real-Time PCR System according to the manufacturer’s protocol. The initial PCR step consisted of a 10 min hold at 95 °C, followed by 40 cycles consisting of 15 s denaturation at 95 °C and 1 min annealing/extension at 60 °C. The expression levels of miR-29a (Assay ID: 002112), miR-29b (Assay ID: 000413) and miR-29c (Assay ID: 000587) were analysed by TaqMan quantitative real-time PCR (TaqMan MicroRNA Assay; Applied Biosystems) and normalised to RNU48 (Assay ID: 001006). TaqMan probes and primers for LAMC2 (P/N: Hs01043711_m1), ITGA6 (P/N: Hs01041011_m1) and GUSB (P/N: Hs99999908_m1) as an internal control were obtained from Applied Biosystems (Assay-On-Demand Gene Expression Products). The ΔΔCt method was adopted and applied to calculate the relative quantities of subject genes. All reactions were performed in triplicate, and included negative control reactions that lacked cDNA.
Mature miRNA transfection and small interfering RNA treatment
The following mature miRNAs species were used in this study: mirVana miRNA mimic for hsa-miR-29a-3p (product ID: MC12499), hsa-miR-29b-3p (product ID: MC10103) and hsa-miR-29c-3p (product ID: MC10518) (Applied Biosystems). The following siRNAs were used: Stealth Select RNAi siRNA; si-LAMC2 (Cat no. HSS105965), si-ITGA6 (Cat no. HSS179959) (Invitrogen) and negative control miRNA/siRNA (P/N: AM17111, Applied Biosystems). The RNAs were incubated with OPTI-MEM (Invitrogen) and Lipofectamine RNAiMax reagent (Invitrogen) as described previously (Ichimi et al, 2009). Transfection efficiency of miRNA in cell lines was confirmed based on downregulation of TWF1 (PTK9) mRNA following transfection with miR-1 as previously reported (Ichimi et al, 2009).
Cell proliferation, migration and invasion assays
Cells were transfected with 10 nM miRNA or siRNA by reverse transfection and plated in 96-well plates at 3 × 103 cells per well. After 72 h, cell proliferation was determined with the XTT assay, using the Cell Proliferation Kit II (Roche Molecular Biochemicals, Mannheim, Germany) as previously reported (Chiyomaru et al, 2010).
A cell migration assay was performed using BD Falcon Cell Culture Inserts (BD Biosciences, Franklin Lakes, NJ, USA) that contained uncoated Transwell polycarbonate membrane filters with 8 μm pores in 24-well tissue culture plates. Cells were transfected with 10 nM miRNA or siRNA by reverse transfection and plated in 10 cm dishes at 8 × 105 cells. After 48 h, the cells were collected and 2 × 105 cells were added to the upper chamber of each migration well and were allowed to migrate for 48 h. After gentle removal of the nonmigratory cells from the filter surface of the upper chamber, the cells that migrated to the lower side were fixed and stained with Diff-Quick (Sysmex Corporation, Kobe, Japan). The number of cells that migrated to the lower surface was determined microscopically by counting four areas of constant size per well.
A cell invasion assay was carried out using modified Boyden chambers containing Transwell-precoated Matrigel membrane filter inserts with 8 μm pores in 24-well tissue culture plates at 2 × 105 cells per well (BD Biosciences). All experiments were performed in triplicate.
In silico analysis, genome-wide gene expression analysis and miR-29s target search
To identify miR-29s target genes, we used in silico analysis and genome-wide gene expression analysis. First, we screened genes using TargetScan Release 6.2 (http://www.targetscan.org/). These genes were then categorised into KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways using GENECODIS analysis (Tabas-Madrid et al, 2012) (http://genecodis.cnb.csic.es/). To identify upregulated genes in HNSCC, we analysed a publicly available gene expression data set in GEO (Accession number: GSE9844). To gain further insight into which genes were affected by miR-29s, we performed genome-wide gene expression analysis using miR-29a transfection of SAS. SurePrint G3 Human GE 8 × 60K Microarray (Agilent Technologies) was used for expression profiling of miR-29a transfectants in comparison with negative control miRNA transfectants.
Western blotting
Cells were harvested 72 h after transfection and lysates were prepared. From each lysate, 50 μg of protein was separated on a Mini-PROTEAN TGX gel (Bio-Rad, Hercules, CA, USA) and transferred to PVDF membranes. Immunoblotting was performed with mouse LAMC2 antibody (1 : 500, MAB19562, Merck Millipore, Billerica, MA, USA) and rabbit ITGA6 antibody (1 : 1000, ab97760, Abcam, Cambridge, UK) with GAPDH antibody (1 : 1000, ab8245, Abcam) used as an internal control. The membrane was washed and incubated with anti-mouse IgG HRP-linked antibody (7076, Cell Signaling Technology, Danvers, MA, USA) or anti-rabbit IgG HRP-linked antibody (7074, Cell Signaling Technology). Complexes were visualised with an Immun-Star WesternC Chemiluminescence Kit (Bio-Rad), and the expression levels of these genes were evaluated by ImageJ software (ver.1.44; http://rsbweb.nih.gov/ij/).
Plasmid construction and dual-luciferase reporter assay
The partial wild-type sequences of the LAMC2 3′-untranslated region (3′-UTR) or those with deleted miR-29s target sites (positions 611–627 and 1430–1436 of LAMC2 3′-UTR) were inserted between the XhoI–PmeI restriction sites in the 3′-UTR of the hRluc gene in psiCHECK-2 vector (C8021; Promega, Madison, WI, USA). The same was done with the partial wild-type sequences of the ITGA6 3′-UTR or those with deleted miR-29s target site (positions 1570–1576 of ITGA6 3′-UTR). The sequences of the oligonucleotides were described in the Supplementary Document. The SAS cells were transfected with 5 ng of vector and 10 nM of miR-29s using Lipofectamine 2000 (Invitrogen). The activities of firefly and Renilla luciferases in cell lysates were determined with a dual-luciferase assay system (E1910; Promega). Normalised data were calculated as the quotient of Renilla/firefly luciferase activities.
Statistical analysis
The relationships between two groups and the numerical values obtained by real-time RT–PCR were analysed using the paired t-test. Spearman’s rank test was used to evaluate the correlation between the expressions of miR-29s. The relationship among more than three variables and numerical values were analysed using the Bonferroni adjusted Mann–Whitney U-test. All analyses were performed using Expert StatView (version 4, SAS Institute Inc., Cary, NC, USA).
Results
Expression of miR-29s in clinical HNSCC specimens
The chromosomal locations of miR-29s in the human genome are shown in Supplementary Figure 1. These miRNAs are clustered at two different human genomic loci, miR-29b-1 and miR-29a at 7q32.3 and miR-29b-2 and miR-29c at 1q32.2.
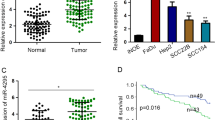
To validate our past miRNA profiling results, we evaluated miR-29s expression in 23 clinical HNSCC specimens. The expression levels of miR-29a, miR-29b and miR-29c were significantly lower in tumour tissues than in corresponding adjacent noncancerous epithelia (P=0.029, P=0.034 and P<0.0001, respectively, Figure 1A). Spearman’s rank test showed positive correlation between the expression of miR-29a and that of miR-29b (R=0.55 and P=0.0001, Figure 1B). Similarly, the expression of miR-29c was positively correlated with that of miR-29b (R=0.62 and P<0.0001, Figure 1B).

Expression levels of miR-29s in HNSCC clinical specimens. (A) Expression levels of miR-29a, miR-29b and miR-29c in HNSCC clinical specimens were measured by qRT–PCR. The RNU48 was used for normalisation. (B) Correlations between miR-29a-miR-29b and miR-29b-miR-29c were determined in HNSCC specimens.
Effect of restoring miR-29s on cell proliferation, migration and invasion activities in HNSCC cell lines
To investigate the functional effects of miR-29s, we performed gain-of-function studies using miRNA transfection of SAS and FaDu cell lines. The XTT assay demonstrated that cell proliferation was significantly inhibited in miR-29s transfectants in comparison with the mock or miR-control transfectant cells. Specifically, we observed the following growth, expressed as a percentage of the mock: (1) SAS–Mock, 100.0±2.4; miR-control, 110.8±4.9; miR-29a, 48.6±4.2; miR-29b, 56.4±5.0; miR-29c, 60.7±4.9; (2) FaDu–Mock, 100.0±1.9; miR-control, 104.5±2.6; miR-29a, 82.3±4.4; miR-29b, 84.5±1.8; miR-29c, 62.7±0.7, with P<0.005 for both (Figure 2A).

Effect of miR-29s transfection on cell proliferation, migration and invasion in SAS and FaDu. (A) Cell proliferation 72 h after transfection with miR-29s (10 nM) was determined with the XTT assay. (B) Cell migration activity 48 h after transfection with miR-29s (10 nM) was determined with the migration assay. (C) Cell invasion activity 48 h after transfection with miR-29s (10 nM) was determined with the Matrigel invasion assay. *P<0.005.
The migration assay demonstrated that cell migration activity was significantly inhibited in miR-29s transfectants in comparison with the mock or miR-control transfectant cells. Specifically, we observed the following migration activities, expressed as a percentage of the mock: (1) SAS–Mock, 100.0±5.3; miR-control, 100.2±4.8; miR-29a, 10.2±1.3; miR-29b, 7.9±0.9; miR-29c, 5.2±0.8; (2) FaDu–Mock, 100.0±8.7; miR-control, 113.4±13.8; miR-29a, 8.3±4.4; miR-29b, 14.0±4.0; miR-29c, 8.6±2.3, with P<0.005 for both (Figure 2B).
The Matrigel invasion assay demonstrated that cell invasion activity was significantly inhibited in miR-29s transfectants in comparison with the mock or miR-control transfectant cells. Specifically, we observed the following migration activities, expressed as a percentage of the mock: (1) SAS–Mock, 100.0±13.6; miR-control, 99.2±11.4; miR-29a, 11.1±2.8; miR-29b, 6.9±1.8; miR-29c, 6.4±1.8 and (2) FaDu—Mock, 100.0±16.3; miR-control, 94.9±4.4; miR-29a, 5.8±1.8; miR-29b, 10.9±2.0; miR-29c, 8.3±2.2, with P<0.005 for both (Figure 2C).
Selection of candidate genes targeted by miR-29s
To identify genes targeted by miR-29s, we used in silico analysis and genome-wide gene expression analysis. Our strategy for selection of miR-29s-targeted genes is shown in Supplementary Figure 2. First, we screened miR-29s-targeted genes using the TargetScan database and identified 2627 genes (Supplementary Table 2). These genes were then categorised into KEGG pathways using GENECODIS analysis and 83 pathways were identified as significantly enriched pathways (Supplementary Table 3). Among these pathways, we focussed on the focal adhesion pathway because this pathway was implicated in cancer cell migration and invasion. A total of 58 genes were identified in this pathway (Supplementary Table 4). The gene set was then analysed with a publicly available gene expression data set in GEO (accession number: GSE9844) and genes upregulated (log2 ratio >0.5) in HNSCC were chosen. To gain further insight into which genes were affected by miR-29s, we performed genome-wide gene expression analysis using SAS, and genes downregulated (log2 ratio<−0.5) by miR-29a transfection were selected. Entries from the gene expression data were approved by GEO, and were assigned GEO accession number GSE47657. As a result, 16 candidate genes were identified as miR-29s targets in the focal adhesion pathway (Table 1). Among those candidates, we focussed on LAMC2 (a component of laminin-332), which is a specific ligand of ITGA6 (a component of α6β4 integrin). Laminin 332–α6β4 integrin interaction is known to promote tumour invasion and cell survival through PI3K and RAC1 activation. Therefore, we focussed on LAMC2 and ITGA6 in further analyses.
LAMC2 and ITGA6 were directly regulated by miR-29s
We performed qRT–PCR and western blotting in SAS to investigate whether LAMC2 and ITGA6 expression was downregulated by restoration of miR-29s. The mRNA and protein expression levels of LAMC2/LAMC2 were significantly repressed in miR-29s transfectants in comparison with mock or miR-control transfectants (P<0.005, Figure 3A and B). Similarly, the mRNA and protein expression levels of ITGA6/ITGA6 were significantly repressed in miR-29s transfectants (P<0.005, Figure 4A and B).

Direct regulation of LAMC2 by miR-29s. (A) The LAMC2 mRNA expression 72 h after transfection with 10 nM miR-29s in SAS was evaluated with qRT–PCR. As an internal control, GUSB was used. **P<0.005. (B) LAMC2 protein expression 72 h after transfection with miR-29s in SAS was evaluated with western blot analysis. As a loading control, GAPDH was used. The expression ratio of LAMC2/GAPDH was calculated with ImageJ software. (C) TargetScan database analysis indicated two putative miR-29s-binding sites existed in the LAMC2 3′-UTR (positions 611–627 and 1430–1436). With regard to these two sites, partial wild-type sequences of LAMC2 3′-UTR and those with the deleted miR-29s target sites were constructed and inserted between the XhoI and PmeI restriction sites in the 3′-UTR of the hRluc gene in the psiCHECK-2 vector. (D) The vectors were co-transfected with miR-29s or miR-control into SAS cells. Firefly luciferase activity was normalised to Renilla luciferase activity. Relative luciferase activity in miR-29s transfectants was compared with that in miR-control cultures, which was set at 1, in cells transfected with wild type 3′-UTR or deletion 3′-UTR. *P<0.0083.

Direct regulation of ITGA6 by miR-29s. (A) The ITGA6 mRNA expression 72 h after transfection with 10 nM miR-29s in SAS was evaluated with qRT–PCR. As an internal control, GUSB was used. **P<0.005. (B) ITGA6 protein expression 72 h after transfection with miR-29s in SAS was evaluated with western blot analysis. As a loading control, GAPDH was used. The expression ratio of ITGA6/GAPDH was calculated with ImageJ software. (C) TargetScan database analysis indicated a putative miR-29s-binding site existed in the ITGA6 3′-UTR (positions 1570–1576). With regard to this site, partial wild-type sequences of ITGA6 3′-UTR and those with the deleted miR-29s target site were constructed and inserted between the XhoI and PmeI restriction sites in the 3′-UTR of the hRluc gene in psiCHECK-2 vector. (D) The vectors were co-transfected with miR-29s or miR-control into SAS cells. Firefly luciferase activity was normalised to Renilla luciferase activity. Relative luciferase activity in miR-29s transfectants was compared with that in miR-control cultures, which was set at 1, in cells transfected with wild-type 3′UTR or deletion 3′-UTR. *P<0.0083.
We performed a luciferase reporter assay in SAS to determine whether LAMC2 and ITGA6 mRNA had target sites for miR-29s. The TargetScan database predicted that two putative miR-29s-binding sites existed in LAMC2 3′-UTR (positions 611–627 and 1430–1436, Figure 3C). We used vectors encoding either the partial wild-type sequence of the 3′-UTR of LAMC2 mRNA, including the predicted miR-29s target sites, or ‘deletion’ vectors, that is, those lacking the miR-29s target sites. We found that the luminescence intensity was significantly reduced by transfection of miR-29s with the vector carrying the wild-type 3′-UTR of LAMC2, whereas transfection with deletion vectors (positions 611–627 or 1430–1436 had been removed) blocked the decrease in luminescence (P<0.0083, Figure 3D). As for ITGA6, the TargetScan database predicted a putative miR-29s-binding site in ITGA6 3′-UTR (positions 1570–1576). We constructed a vector encoding either the partial wild-type sequences of 3′-UTR of ITGA6 or those with a deleted miR-29s target site (Figure 4C). The luminescence intensity was significantly reduced by transfection of miR-29s with the vector of wild-type 3′-UTR of LAMC2 (P<0.0083, Figure 4D). Although the luminescence intensity was also reduced by transfection of miR-29s with the deletion vector, the reduction was smaller than that with wild type (Figure 4D).
Effects of silencing LAMC2 and ITGA6 on cell proliferation, migration and invasion in SAS
To investigate the functional role of LAMC2 and ITGA6, we performed loss-of-function studies using si-LAMC2 and si-ITGA6 transfectants. First, we evaluated the knockdown efficiency of si-LAMC2 and si-ITGA6 treatments in SAS. Western blotting and qRT–PCR indicated that the two siRNAs effectively downregulated ITGA6 and LAMC2 expression in SAS (Supplementary Figure 3).
The XTT assays demonstrated that cell proliferation was significantly inhibited in si-LAMC2 transfectants in comparison with the mock or si-control transfectant in SAS (percentage of the mock: 64.7±2.2, with P<0.0167; Figure 5A). On the other hand, proliferation was not inhibited in si-ITGA6 transfectants in comparison with the mock or si-control transfectants in SAS (Figure 5B).

Effect of si- LAMC2 or si- ITGA6 transfection on cell proliferation, migration and invasion in SAS. Cell proliferation 72 h after transfection with 10 nM si-LAMC2 (A) or si-ITGA6 (B) in SAS was determined with the XTT assay. Cell migration activity 48 h after transfection with 10 nM si-LAMC2 (C) or si-ITGA6 (D) in SAS was determined with the migration assay. Cell invasion activity 48 h after transfection with 10 nM si-LAMC2 (E) or si-ITGA6 (F) in SAS was determined with the Matrigel invasion assay. *P<0.0167.
The migration assays demonstrated that cell migration activity was significantly inhibited in both si-LAMC2 and si-ITGA6 transfectants in comparison with the mock or si-control transfectants in SAS (percentage of the mock: si-LAMC2, 14.3±7.2; si-ITGA6, 15.8±12.5, with P<0.0167 for both; Figure 5C and D).
Matrigel invasion assays demonstrated that cell invasion activity was significantly inhibited in both si-LAMC2 and si-ITGA6 transfectants in comparison with the mock or si-control transfectants in SAS (percentage of the mock: si-LAMC2, 23.4±14.0; si-ITGA6, 19.6±14.3, with P<0.0167 for both; Figure 5E and F).
Discussion
In the postgenome sequencing era, improved understanding of noncoding RNA is necessary for continued progress in cancer research. For elucidation of the molecular mechanisms underlying HNSCC, we have examined tumour-suppressive miRNAs, focussing on their regulated molecular targets and novel cancer pathways based on HNSCC expression signatures (Kikkawa et al, 2010; Nohata et al, 2011). Our previous miRNA expression signatures demonstrated that the miR-29s family (miR-29a/b/c) was reduced in several types of cancers (Kikkawa et al, 2010; Nohata et al, 2011). Decreased expression of miR-29s was described in other types of cancers such as cholangiocarcinoma, nasopharyngeal cancer, non-small cell lung cancer, cervical cancer, hepatocellular carcinoma, malignant peripheral nerve sheath tumour and mantle cell lymphoma (Fabbri et al, 2007; Mott et al, 2010; Xiong et al, 2010; Zhao et al, 2010; Li et al, 2011; Presneau et al, 2013), all of which are consistent with our results. In contrast, upregulation of miR-29s was reported in breast cancer, colon cancer and acute myeloid leukaemia (Gebeshuber et al, 2009; Han et al, 2010; Fu et al, 2012). Therefore, the molecular mechanisms by which miR-29s expression is deregulated vary according to cancer types. In this study, our data showed that all members of the miR-29s family were significantly reduced in cancer tissues. Moreover, restoration of miR-29s strongly inhibited cancer cell migration and invasion. Thus, the miR-29s family functions as a group of tumour suppressors, and they might contribute to metastasis in HNSCC.
To elucidate the silencing mechanisms of miR-29s in HNSCC cells, we performed preliminary analysis of 5-Aza-dC treatment as a demethylation agent on HNSCC cells. Our data demonstrated that miR-29s were not recovered by the treatment (data not shown), suggesting that silencing of miR-29s were not caused by methylation of CpG islands on their genomic regions. Three annotated genes (CR1L, CD46 and C1orf32) are located around the miR-29b-2/miR-29c cluster region on human chromosome 1q32.2 (Supplementary Figure 1). Our microarray expression data of HNSCC (GEO accession number: GSE 36951) showed that expression changes of the three genes were not observed in cancer tissues. According to the UniGene database, expression levels of other genes shown in Supplementary Figure 1 are extremely low.
Although the molecular mechanism by which miR-29s is silenced in HNSCC is still unknown, some important studies have examined the promoter region of miR-29s in the human genome. The human miR-29b-1/miR-29a promoter region contains two putative E-box sites (MYC-binding site), a Gli-binding site and four NF-κB-binding sites. Increased expression of MYC silenced miR-29b-1/miR-29a expression in cholangiocarcinoma cells (Mott et al, 2010). Furthermore, NF-κB signalling, which is known to be activated in inflammation-related cancers, directly repressed miR-29b-1/miR-29a promoter activity (Wang et al, 2008; Mott et al, 2010). Recent data showed that GATA3, a transcription factor, promotes differentiation, suppresses metastasis and alters the tumour microenvironment in breast cancer by inducing miR-29b expression (Chou et al, 2013). Thus, it will be necessary to identify the transcription factors contributing to HNSCC. Further detailed analysis is needed to understand the molecular mechanisms of miR-29s silencing in HNSCC cells.
Metastasis is the final stage of cancer progression and is the cause of most cancer deaths. The epithelial-to-mesenchymal transition (EMT) is considered to be an important step in cancer progression and metastasis, and TGF-β signalling contributes to EMT processes (Heldin et al, 2012). Recent data have suggested that TGF-β1 inhibits the expression of miR-29s and promotes the expression of extracellular matrix (ECM) components (Maurer et al, 2010; Roderburg et al, 2011). These data suggest that miR-29s could play an important role in modulating TGF-β signalling during cancer cell migration and invasion. It will be of interest to more fully identify the molecular targets and pathways of miR-29s in HNSCC, particularly those contributing to cancer cell migration and invasion.
The miRNAs are unique in their ability to regulate many protein-coding genes. Bioinformatic predictions indicate that miRNAs regulate >30% of protein-coding genes (Lewis et al, 2005). We have taken the position that the identification of novel cancer pathways and responsible genes regulated by tumour-suppressive miR-29s is an important first step in understanding HNSCC oncogenesis. Based on this view, we categorised miR-29s target genes into known pathways using the KEGG pathway database. These data have facilitated the understanding of tumour-suppressive miRNA-regulated molecular pathways in human cancer. We devised this method of analysis and found that tumour-suppressive miRNA could efficiently regulate cancer-associated pathways (Kinoshita et al, 2012; Nohata et al, 2013; Yoshino et al, 2013).
In this study, we focussed on the ‘focal adhesion’ pathway because restoration of miR-29s inhibited cancer cell migration and invasion in HNSCC cell lines. Cell adhesion is the binding of a cell to a surface, such as an ECM or another cell, and a fundamental determinant of a wide range of cell biology responses and signalling (Parsons et al, 2010). Recent evidences suggested that several miRNAs including miR-29s regulated multiple molecular components of the cell adhesion machinery and contributed to functional key regulators of adhesion-associated processes (Valastyan and Weinberg, 2011). Our hypothesis is that downregulation of tumour-suppressive miRNAs in cancer cells leads to upregulation of oncogenes. Therefore, we combined the gene expression data of miR-29a transfectants and the GEO expression data of upregulated genes in HNSCC, generating 16 candidate target genes for miR-29s in focal adhesion pathways. We focussed on laminin γ2 (LAMC2), a member of laminin-332, and α6 integrin (ITGA6) as responsible genes that contribute to cancer cell migration and invasion in HNSCC. Numerous clinical studies indicated that overexpression of LAMC2 was positively correlated with invasiveness and poor survival in several types of cancer (Guess and Quaranta, 2009), suggesting that LAMC2 functions as an oncogene and deeply contributes to cancer metastasis. Because of the small number of the HNSCC samples, no positive data were obtained between clinical significance of the HNSCC patients and the LAMC2 and ITGA6 expression levels in the present study. Further examination should be necessary to elucidate the expression levels of LAMC2 and ITGA6 and the clinicopathological parameters and patient survival of HNSCC.
Integrins are a large family of cell surface receptors composed of two subunits (α and β) that bind to ECM components. Most types of cells require integrin-mediated signal pathways for proliferation, migration, invasion and survival. To date, a total of 18 different α-subunits and 8 different β-subunits have been identified, accounting for at least 24 distinct integrin heterodimers (Gilcrease, 2007). Among these integrins, laminin-332 interacts with two major integrin receptors, α3β1 and α6β4, promoting the formation of focal adhesions and stable anchoring contacts (Marinkovich, 2007). The α6β4 integrin is reportedly involved as a link between the cytoskeleton and ECM as well as in the activation of a variety of intercellular signalling processes in cooperation with growth factor receptors (Falcioni et al, 1997; Mariotti et al, 2001; Trusolino et al, 2001).
Of particular interest, laminin-332 and α6β4 integrin interaction triggers a number of signalling cascades in cancer cells, promoting both cell migration and cancer cell survival (Marinkovich, 2007). Furthermore, our previous data demonstrated that tumour-suppressive miR-218, which was frequently reduced in HNSCC, directly regulated laminin-332, suggesting that miR-218 contributed to cancer cell migration and invasion through regulating focal adhesion pathways (Kinoshita et al, 2012). Thus, the available data suggest that tumour-suppressive miR-29s and miR-218 contribute to cancer cell migration and invasion through their regulation of focal adhesion pathways, especially targeting laminin-332 and α6β4 integrin signalling (Figure 6). Evidence indicates that integrins crosstalk with receptor tyrosine kinases in an integrin type-dependent manner through a variety of specific mechanisms (Falcioni et al, 1997; Mariotti et al, 2001; Trusolino et al, 2001). Thus, the understanding of tumour-suppressive miRNAs (miR-29s and miR-218) and their regulation of laminin–integrin signalling should shed light on HNSCC metastasis as well as delineate more effective strategies for future therapeutic interventions for this disease.

Illustration of tumour-suppressive miR-29s and miR-218 regulation of laminin-332 and α 6 β 4 integrin. Regulation of laminin-332 and α6β4 integrin by miR-29s and miR-218 is shown (modified a Figure in Guess and Quaranta, 2009). Laminin-332, which consists of laminin α3, β3 and γ2 chains, is a major component of basement membranes, but is also an important autocrine ligand produced by cancer cells to promote tumourigenesis. Laminin-332–α6β4 integrin interaction promotes tumour invasion and survival by activation of PI3K and RAC1. Both LAMC2 and ITGA6 were directly regulated by tumour-suppressive miR-29s, whereas LAMB3 was directly regulated by tumour-suppressive miR-218 (our previous study). In addition, ITGB4 has a putative miR-218-binding site in the 3′-UTR region.
Conclusions
Our data showed that all members of the miR-29s group were frequently downregulated in HNSCC. They function as tumour suppressors and normally inhibit cancer cell migration and invasion through their regulation of focal adhesion pathways, especially via laminin-332 and α6β4 integrin. Elucidation of cancer pathways regulated by tumour-suppressive miR-29s should shed light on HNSCC metastasis as well as delineate more effective strategies for future therapeutic interventions for this disease.
Change history
12 November 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Avissar M, Christensen BC, Kelsey KT, Marsit CJ (2009) MicroRNA expression ratio is predictive of head and neck squamous cell carcinoma. Clin Cancer Res 15: 2850–2855.
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297.
Chiyomaru T, Enokida H, Tatarano S, Kawahara K, Uchida Y, Nishiyama K, Fujimura L, Kikkawa N, Seki N, Nakagawa M (2010) miR-145 and miR-133a function as tumour suppressors and directly regulate FSCN1 expression in bladder cancer. Br J Cancer 102: 883–891.
Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z (2013) GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol 15: 201–213.
Esquela-Kerscher A, Slack FJ (2006) Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 6: 259–269.
Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 104: 15805–15810.
Falcioni R, Antonini A, Nistico P, Di Stefano S, Crescenzi M, Natali PG, Sacchi A (1997) Alpha 6 beta 4 and alpha 6 beta 1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp Cell Res 236: 76–85.
Fu J, Tang W, Du P, Wang G, Chen W, Li J, Zhu Y, Gao J, Cui L (2012) Identifying microRNA-mRNA regulatory network in colorectal cancer by a combination of expression profile and bioinformatics analysis. BMC Syst Biol 6: 68–0509-6-68.
Gebeshuber CA, Zatloukal K, Martinez J (2009) miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO J Rep 10: 400–405.
Gilcrease MZ (2007) Integrin signaling in epithelial cells. Cancer Lett 247: 1–25.
Guess CM, Quaranta V (2009) Defining the role of laminin-332 in carcinoma. Matrix Biol 28: 445–455.
Han YC, Park CY, Bhagat G, Zhang J, Wang Y, Fan JB, Liu M, Zou Y, Weissman IL, Gu H (2010) microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med 207: 475–489.
Heldin CH, Vanlandewijck M, Moustakas A (2012) Regulation of EMT by TGFbeta in cancer. FEBS Lett 586: 1959–1970.
Hui AB, Lenarduzzi M, Krushel T, Waldron L, Pintilie M, Shi W, Perez-Ordonez B, Jurisica I, O'Sullivan B, Waldron J, Gullane P, Cummings B, Liu FF (2010) Comprehensive MicroRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res 16: 1129–1139.
Ichimi T, Enokida H, Okuno Y, Kunimoto R, Chiyomaru T, Kawamoto K, Kawahara K, Toki K, Kawakami K, Nishiyama K, Tsujimoto G, Nakagawa M, Seki N (2009) Identification of novel microRNA targets based on microRNA signatures in bladder cancer. Int J Cancer 125: 345–352.
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60: 277–300.
Kariya Y, Kariya Y, Gu J (2009) Roles of laminin-332 and alpha 6 beta 4 integrin in tumor progression. Mini Rev Med Chem 9: 1284–1291.
Kikkawa N, Hanazawa T, Fujimura L, Nohata N, Suzuki H, Chazono H, Sakurai D, Horiguchi S, Okamoto Y, Seki N (2010) miR-489 is a tumour-suppressive miRNA target PTPN11 in hypopharyngeal squamous cell carcinoma (HSCC). Br J Cancer 103: 877–884.
Kinoshita T, Hanazawa T, Nohata N, Kikkawa N, Enokida H, Yoshino H, Yamasaki T, Hidaka H, Nakagawa M, Okamoto Y, Seki N (2012) Tumor suppressive microRNA-218 inhibits cancer cell migration and invasion through targeting laminin-332 in head and neck squamous cell carcinoma. Oncotarget 3: 1386–1400.
Kriegel AJ, Liu Y, Fang Y, Ding X, Liang M (2012) The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol Genomics 44: 237–244.
Leemans CR, Braakhuis BJ, Brakenhoff RH (2011) The molecular biology of head and neck cancer. Nat Rev Cancer 11: 9–22.
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20.
Li Y, Wang F, Xu J, Ye F, Shen Y, Zhou J, Lu W, Wan X, Ma D, Xie X (2011) Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPV-related target genes for miR-29. J Pathol 224: 484–495.
Marinkovich MP (2007) Tumour microenvironment: laminin 332 in squamous-cell carcinoma. Nat Rev Cancer 7: 370–380.
Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, Giancotti FG (2001) EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J Cell Biol 155: 447–458.
Mattick JS (2004) RNA regulation: a new genetics? Nat Rev Genet 5: 316–323.
Maurer B, Stanczyk J, Jungel A, Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE, Michel BA, Distler JH, Gay S, Distler O (2010) MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum 62: 1733–1743.
Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, Fernandez-Zapico ME (2010) Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem 110: 1155–1164.
Nohata N, Hanazawa T, Kikkawa N, Sakurai D, Fujimura L, Chiyomaru T, Kawakami K, Yoshino H, Enokida H, Nakagawa M, Katayama A, Harabuchi Y, Okamoto Y, Seki N (2011) Tumour suppressive microRNA-874 regulates novel cancer networks in maxillary sinus squamous cell carcinoma. Br J Cancer 105: 833–841.
Nohata N, Hanazawa T, Kinoshita T, Inamine A, Kikkawa N, Itesako T, Yoshino H, Enokida H, Nakagawa M, Okamoto Y, Seki N (2013) Tumour-suppressive microRNA-874 contributes to cell proliferation through targeting of histone deacetylase 1 in head and neck squamous cell carcinoma. Br J Cancer 108: 1648–1658.
Parsons JT, Horwitz AR, Schwartz MA (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11: 633–643.
Presneau N, Eskandarpour M, Shemais T, Henderson S, Halai D, Tirabosco R, Flanagan AM (2013) MicroRNA profiling of peripheral nerve sheath tumours identifies miR-29c as a tumour suppressor gene involved in tumour progression. Br J Cancer 108: 964–972.
Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, Tacke F, Trautwein C, Luedde T (2011) Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53: 209–218.
Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P (2008) MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci USA 105: 5874–5878.
Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A (2012) GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res 40: W478–W483.
Trusolino L, Bertotti A, Comoglio PM (2001) A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 107: 643–654.
Valastyan S, Weinberg RA (2011) Roles for microRNAs in the regulation of cell adhesion molecules. J Cell Sci 124: 999–1006.
Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, Cheng A, Hall BM, Qualman SJ, Chandler DS, Croce CM, Guttridge DC (2008) NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14: 369–381.
Wang Y, Zhang X, Li H, Yu J, Ren X (2013) The role of miRNA-29 family in cancer. Eur J Cell Biol 92: 123–128.
Xiong Y, Fang JH, Yun JP, Yang J, Zhang Y, Jia WH, Zhuang SM (2010) Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 51: 836–845.
Yoshino H, Enokida H, Itesako T, Tatarano S, Kinoshita T, Fuse M, Kojima S, Nakagawa M, Seki N (2013) Epithelial-mesenchymal transition-related microRNA-200s regulate molecular targets and pathways in renal cell carcinoma. J Hum Genet 58: 508–516.
Zhao JJ, Lin J, Lwin T, Yang H, Guo J, Kong W, Dessureault S, Moscinski LC, Rezania D, Dalton WS, Sotomayor E, Tao J, Cheng JQ (2010) microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood 115: 2630–2639.
Acknowledgements
This work was supported by KAKENHI Grant numbers 23592505, 24592590 and 25861528.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kinoshita, T., Nohata, N., Hanazawa, T. et al. Tumour-suppressive microRNA-29s inhibit cancer cell migration and invasion by targeting laminin–integrin signalling in head and neck squamous cell carcinoma. Br J Cancer 109, 2636–2645 (2013). https://doi.org/10.1038/bjc.2013.607
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.607
Keywords
This article is cited by
-
The tumor ecosystem in head and neck squamous cell carcinoma and advances in ecotherapy
Molecular Cancer (2023)
-
MiR-29c-3p/C1QTNF6 Restrains the Angiogenesis and Cell Proliferation, Migration and Invasion in Head and Neck Squamous Cell Carcinoma
Molecular Biotechnology (2023)
-
Integrin α6 overexpression promotes lymphangiogenesis and lymphatic metastasis via activating the NF-κB signaling pathway in lung adenocarcinoma
Cellular Oncology (2022)
-
LncRNA EBLN3P promotes the progression of osteosarcoma through modifying the miR-224-5p/Rab10 signaling axis
Scientific Reports (2021)
-
LncRNA HULC induces the progression of osteosarcoma by regulating the miR-372-3p/HMGB1 signalling axis
Molecular Medicine (2020)
