
Abstract
The discovery of unprecedented, non-native phases of transition metal oxides is a fascinating theme in materials chemistry, including the development of new synthetic strategies with excellent size- and phase-selectivity, and elucidation of their unique physicochemical properties. Most of the first-row transition metal monoxides, such as MnO, FeO, CoO and NiO, are known to exist in a cubic phase, but only ZnO adopts a hexagonal structure. This review summarizes recent results on the syntheses and characterization of the non-native phases of h-CoO, h-MnO and c-ZnO transition metal monoxides, compared to their native phases. The investigation of the size- and phase-selective synthetic methods, formation mechanism and unique physicochemical properties of non-native transition metal monoxides would have an enormous impact on materials chemistry and open a promising avenue for magnetic, electronic and spintronic applications.
Similar content being viewed by others

Introduction
First-row transition metal monoxides are an important group of materials because they are thermodynamically and chemically stable, form a wide variety of crystal structures and display many interesting physicochemical properties that are useful for numerous potential applications such as catalysis, energy storage, magnetic data storage, sensors, non-linear optics, electrode materials and ferrofluids.1, 2, 3, 4, 5 In particular, these transition metal monoxides adopt two simple crystal structures, cubic and hexagonal phases. The cubic close-packed (ccp) structure, where cations occupy the octahedral sites of the ccp array of anions, is a stable form in most metal monoxides such as MnO, FeO, CoO and NiO.6, 7, 8 On the other hand, the hexagonal close-packed (hcp) structure, where cations are located at tetrahedral sites of the hcp array of anions, is thermodynamically unstable relative to the ccp structure, although the hcp structure exists in many of inorganic binary materials such as ZnO, metal sulfides, and selenides.9, 10 To extend the range of available metal monoxides, it is extremely important to discover size- and phase-selective synthetic methods that can provide non-native, unprecedented transition metal monoxides and to explore their unique physicochemical properties.
A phase that is thermodynamically less stable is hardly observed in bulk forms. However, in nanomaterials, the surface energy exceeds the crystal formation energy, which stabilizes an unstable phase of the nano-structured materials and dominates all of their physicochemical properties.11, 12, 13, 14 Importantly, thermodynamically unstable phases can be stabilized by coupling the excess surface energy of nanomaterials with careful control of kinetic parameters in synthetic reactions. For instance, the cubic phase of MnSe is generally stable in bulk powders and thin films, but the hexagonal phase is formed in the nanometer range.15, 16 In some cases, these non-native crystal phases exhibit superior physicochemical properties that are uniquely suitable for technological applications. The hexagonal structure, however, does not have a center of inversion symmetry and thus permits useful physicochemical properties such as piezoelectricity, pyroelectricity, second-harmonic generation and other interesting phenomena.17, 18
This review focuses on the recent studies of the syntheses and physicochemical characterization of the non-native phases, as well as the comparison of their physicochemical properties with those of the native phase in the first-row transition metal monoxides. Detailed discussions include size-selective and phase-controlled synthetic methods, mechanistic pathways, phase transition, and phase-dependent physicochemical properties of the first-row transition metal monoxides.
Crystal structures of transition metal monoxides
Among numerous transition metal oxides, transition metal monoxides attract interests for all the usual reasons such as stoichiometric preparation, investigation of their surface structures and physicochemical properties, and useful applications.6 In particular, transition metal monoxides are ideal models for the direct verification of the relationship between antiferromagnetic arrangement of atomic spins and crystal distortions.8 Crystal structures of transition metal monoxides are summarized in Figure 1. In particular, the first-row transition metals form stable monoxides, such as TiO, VO, MnO, FeO, CoO, NiO, CuO and ZnO, but reliable evidence has not been found on the existence of ScO and CrO.6 A simplified approach to predicting coordination numbers in binary compounds has employed the relative sizes of the constituent atoms or ions, r+/r-. Smaller cations in the close-packed structure often fit in the tetrahedral holes of the anion lattice, but larger cations fit in the octahedral holes.7 As shown in Figure 1, most of the first-row transition metal monoxides adopt a stable cubic phase with metal ions in octahedral holes.8 A number of studies have focused on the syntheses and characteristic physicochemical properties of stable cubic phases and their potential applications. For example, electronic properties vary depending on the transition metal: TiO and VO have metallic properties, but MnO, FeO, CoO, NiO, CuO and ZnO are in the category of semiconductors.6 Furthermore, in order to extend the range of available metal oxides, it is very important to discover unprecedented, non-native phases. Since Redman and Steward reported a hexagonal CoO phase (h-CoO) in 1962 by observation of additional reflection lines in addition to those of the cubic CoO phase (c-CoO) in X-ray diffraction (XRD),19 several research groups have intensively investigated phase-selective synthetic methods and physicochemical properties of h-CoO nanomaterials.20, 21, 22, 23, 24, 25, 26, 27, 28 After h-CoO was first reported, no new hexagonal metal oxide structure was discovered for five decades. Recently, Nam et al.29 have successfully prepared unprecedented h-MnO by kinetic reaction control.29, 30 The structure of non-native h-MnO has been identified by XRD, and h-MnO nanocrystals are found to exhibit significant physicochemical characteristics such as a large piezoelectric response and controllable p–n conductivity.31, 32 As a result, two new hexagonal phases, h-CoO and h-MnO, have been recently listed in the crystal structures of transition metal monoxides (Figure 1). ZnO, however, has been known to exist as the only stable metal monoxide possessing a hexagonal phase (h-ZnO), presumably, due to relatively smaller Zn cation, prior to the recent discovery of the hexagonal phases in CoO and MnO systems. The existence of cubic ZnO (c-ZnO) was first discovered by Bates et al. in 1962.33 The h-ZnO transforms into a c-ZnO at high pressures, but h-ZnO is easily recovered from c-ZnO after the pressure release, due to the strong tetrahedral preference of Zn cations. The discovery of c-ZnO resulted in intensive investigations on pressure-dependent behaviors of ZnO in the viewpoints of solid state physics.34, 35, 36, 37 The CoO, MnO and ZnO systems have now multiple crystallographic phases of cubic and hexagonal structures, in which various interests emerge in fundamental themes of materials chemistry such as phase-selective preparation, phase transition between the two phases and phase-dependent physicochemical properties. Thorough understanding of the multiphase metal monoxide systems would open a promising avenue for the discovery of new synthetic strategies of other non-native phases of metal oxides and for the invention of entirely new materials of unique physicochemical properties for various device applications.

Crystal structures of transition metal monoxides. The non-native phases are highlighted in red.
Selective syntheses and formation mechanism of h-CoO and c-CoO
Cobaltous oxide typically crystallizes in two crystal phases; c-CoO (space group Fm3m) with octahedral Co2+ ions and h-CoO (space group P63mc) with tetrahedral Co2+ ions. On the basis of its high chemical stability and intriguing catalytic properties, c-CoO has been widely studied for such potential applications as catalysts, the magnetic data storage devices, solid-state sensors and electrode materials in lithium ion batteries.38 A discovery of the non-native h-CoO phase is significant due to not only fundamental scientific interest in synthetic strategies and mechanisms for new materials but also technological interest in newly emerging unique magnetic and electrochemical properties of these materials.
Selective syntheses of colloidal h-CoO and c-CoO
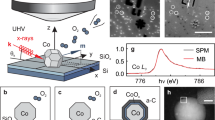
Thermal decomposition of a cobalt precursor under an inert atmosphere yields two distinct phases of h-CoO and c-CoO via kinetic and thermodynamic reaction control, respectively as shown in Scheme 1.23 In general, when two pathways are plausible from a common starting material, one route having a lower activation barrier rapidly affords a product, although this product is energetically less stable than that formed via the other route. In other words, the kinetic control of a reaction generates a thermodynamically less stable product in this case, while the thermodynamic reaction control naturally yields a thermodynamically more stable product. This fundamental concept explains the selective formation of h-CoO and c-CoO nanomaterials by changing the reaction kinetics. Seo et al.20 reported that flash heating of the reaction mixture to 200 °C provides a kinetic reaction condition to produce h-CoO nanocrystals, whereas slow and prolonged heating at 135 °C under a thermodynamic condition yields c-CoO nanocrystals. Similarly, the addition of o-dichlorobenzene (o-DCB) to the reaction mixture changes the reaction condition to a thermodynamic regime.23 In a high concentration of o-DCB, c-CoO nanocrystals are selectively formed, even under synthetic conditions similar to those of h-CoO nanocrystals. Furthermore, adding water to the reaction mixture also converts the reaction conditions into a thermodynamic regime, producing c-CoO nanocrystals in a high yield.39 Figure 2a presents transmission electron microscopy (TEM) and scanning electron microscopy (SEM, in an inset as a hexagonal pyramid) images of h-CoO nanocrystals. The XRD patterns correspond exactly to those of the hexagonal phase with the space group P63mc (Figure 2b). A TEM image and the XRD patterns of c-CoO nanocrystals are presented in Figure 2c and d, respectively. The diffraction peaks of c-CoO are indexed to a cubic structure with the space group Fm3m. The electron energy loss spectroscopy (EELS) for both c-CoO and h-CoO nanocrystals estimated an average atomic ratio (O/Co) of 1.04±0.05 (c-CoO) and 1.01±0.05 (h-CoO), respectively, which approached nearly stoichiometric compositions (Figure 2e).

(a) TEM image and (b) XRD pattern of h-CoO nanoparticles. (c) TEM image and (d) XRD pattern of c-CoO nanoparticles. (e) EELS spectra obtained from Co3O4 (99.995%, Aldrich, St. Louis, MO, USA) and CoO (99.99+%, Aldrich) powders, and prepared c-CoO and h-CoO. The stoichiometry was found to be O/Co=1.34±0.05 for the Co3O4 powders, 1.02±0.05 for the CoO powders, 1.04±0.05 for c-CoO, and 1.01±0.05 for h-CoO, respectively. Adapted from Seo et al.20 and Nam et al.23
Elucidation of selective synthetic mechanism of h-CoO and c-CoO
The selective synthetic mechanism of h-CoO and c-CoO has been investigated by thermal decomposition reactions of the Co(acac)2 precursor in benzylamine. Analyses of the side products of the two reactions shed light on the detailed reaction mechanism.40 In the selective synthesis of h-CoO nanocrystals, four side products are assigned to the reference peaks by gas chromatography-mass spectrometry (GC-MS): benzylamine (1), N-isopropylidenebenzylamine (2), N-benzylacetamide (3) and 4-benzylamino-3-penten-2-one (4).39 Interestingly, compounds 1, 2 and 3 are present in both reactions, but compound 4 is exclusively detected in the selective synthesis of h-CoO nanocrystals, as shown in Scheme 2, indicating the existence of a different synthetic route. On the basis of this observation, independent pathways A and B are proposed for the selective formation of h-CoO nanocrystals (Scheme 2a), which are based on the combination of solvolysis and condensation. Given that the abundances of compounds 2, 3 and 4 are similar, as determined by a GC-MS analysis, the formation of the h-CoO phase proceeds equivalently via reaction pathways A and B. The resulting cobalt hydroxide species undergo condensation to form Co–O–Co bonds, ultimately leading to the generation of CoO seeds. In the selective synthesis of c-CoO nanocrystals in the presence of water, however, the formation of compound 4 is nearly negligible, clearly indicating that pathway B does not occur in this reaction. Instead, pathway C, which involves a C–C bond cleavage of the acac ligand, is proposed as a major and preferred route (Scheme 2b). Under the present basic reaction conditions, hydroxide ions from water are the major species and they attack the carbonyl carbon of the acac ligand, which undergoes a C–C bond cleavage because the hydroxide ion is a more nucleophilic Lewis base than benzylamine. In this pathway, the formation of the carboxylate ligand is a critical step to generate the thermodynamically stable c-CoO phase. It is well known that the carboxylate ligand is tightly bound to the metal center through a strong chelation, which stabilizes metal complexes.41 As a result, decomposition of the cobalt hydroxyl carboxylate intermediate to form CoO becomes slow, and thus, the reaction enters into the regime of thermodynamic control to exclusively yield c-CoO nanocrystals.
Morphology control and growth mechanism of h-CoO nanorods
The crystal structure of h-CoO can be described as a number of alternating planes composed of tetrahedrally coordinated Co2+ and O2− ions, stacked alternatively along the c axis, that is, the [001] direction. The (001) facet on the hexagonal structure has a significantly higher surface energy than those of other facets, leading to the formation of [001]-directed nanorods and nanowires.42 In the selective synthesis of h-CoO nanocrystals, adding o-DCB to the reaction mixture altered the morphology of h-CoO from pyramids to pencil-shaped rods (Figure 3a).23 It is known that a thermodynamic reaction condition generally enhances the growth rate along the [001] direction (Figure 3b) and generates anisotropic nanorods in a stable hexagonal structure.22 Upon the addition of o-DCB to the reaction mixture, the kinetic reaction condition changes to the thermodynamic regime, which leads to elongation of the original h-CoO morphology of pyramids and forms pencil-shaped h-CoO rods.

(a) TEM and (b) HRTEM images of [001]-directed h-CoO nanorods. (c) HRTEM image of [100]-directed Cu-doped h-CoO nanorod and (d) a schematic image of rod-shaped wurtzite nanocrystals. Bottom Table: calculated surface energies of h-CoO and Cu-doped h-CoO facets. ΔE: the change of surface energy by Cu doping. Adapted from Nam et al.23 and Nam et al.44
Several research groups have reported [100]- (or [110]-) directed metal-doped hexagonal nanorods,43 but the explication of an atomistic-level mechanism has remained elusive. In order to address this unknown mechanism, Nam et al.44 prepared [100]-directed Cu-doped h-CoO nanorods, which are more stable compared to the pure h-CoO nanocrystals. The HRTEM image in Figure 3c represents a highly crystalline growth nature of the (100) lattice planes (Figure 3d). The bottom table in Figure 3 summarizes a comparison of the surface energies of pure h-CoO and Cu-doped h-CoO by first-principles calculations. Under the thermodynamic equilibrium conditions, the surface energy of the (001) facet is the highest, followed by the (101), (110) and (100) facets of the h-CoO nanoparticles, and this causes growth of the h-CoO nanocrystals along the [001] direction. In the case of Cu-doped h-CoO nanorods, the surface Cu atoms are greatly preferable to the bulk Cu atoms due to the reduced total energy of surface Cu atoms, and thus the doped Cu atoms mainly substitute Co sites in the top layer of the (001) surface and significantly stabilize the h-CoO structure. This accounts for the interruption of growth of the (001) facet and the growth of (100) facets as metal Cu atoms are doped into the h-CoO nanorods.44 It is notable that the nanorods with a hexagonal phase tend to grow rapidly along the direction parallel to the interrupted plane.43
Phase transition of h-CoO to c-CoO and ultimate formation of spinel-Co3O4
Understanding the relative stability of different crystallographic phases and phase transformations is of great interest in the field of materials chemistry.45, 46 The stability of h-CoO has been investigated in detail for practical applications such as piezoelectric and pyroelectric devices. Jiang et al. reported that the h-CoO phase transformed into the c-CoO phase at 6.9 GPa by means of an in-situ high-pressure synchrotron radiation XRD measurement. The decompression experiments revealed that the h-CoO to c-CoO phase transformation was irreversible, since only the c-CoO phase was detected in the sample after the pressure release.47
The progress of the oxidation reaction of h-CoO has been monitored at regular reaction periods by XRD (Figure 4a–d).23 Interestingly, peaks due to both c-CoO and spinel-Co3O4 simultaneously appear at the early stage of the oxidation reaction, indicating that the phase transition of h-CoO to c-CoO and spinel-Co3O4 takes place rapidly (Figure 4b). The signals due to h-CoO completely disappear within 20 min (Figure 4c), and the oxidation reaction is completed within 5 h to yield pure spinel-Co3O4 nanoparticles with retention of the original morphology of h-CoO nanoparticles (Figure 4d). On the basis of the XRD data, the rearrangements of the cobalt cations and oxygen anions during the oxidation process are clearly understood at an atomic level. The rapid conversion of h-CoO to c-CoO during oxidation is caused by the different rearrangement rates between cobalt cations and oxygen anions. The rearrangement of the oxygen anions in h-CoO is rapid enough to form a ccp array of c-CoO within 20 min, but the cobalt cations that are buried in the ccp array of oxygen anions are too slow to occupy the new positions for the formation of the spinel-Co3O4 structure; this explains the formation of the c-CoO intermediate phase at the initial stage of oxidation.

XRD patterns of h-CoO during oxidation in air: (a) Before and after heat treatment at 240 °C for (b) 10 min, (c) 20 min and (d) 5 h. Adapted from Nam et al.23
Formation of c-Co hollow nanoparallelopipeds from c-CoO nanoparallelopipeds
Cobalt nanomaterials have been widely studied due to their potential applications, mainly in the magnetic data storage.48 Morphology- and structure-related magnetic properties of cobalt nanomaterials have been at the center of scientific issues. Hollow cobalt nanomaterials, however, have rarely been reported, although this morphology is intriguing with regard to unusual magnetic domains and quantum properties.
The c-CoO nanoparallelopipeds in oleylamine undergo a reduction reaction by the oxidation of oleylamine at high temperatures to transform into c-Co hollow nanoparallelopipeds.48 Figure 5 shows the evolution of the morphology of c-Co hollow nanoparallelopipeds with time (Figure 5a–d). Time-dependent XRD patterns during thermolysis of c-CoO show that the signals corresponding to c-Co continuously grow with time and those of c-CoO completely disappear within 3 h (Figure 6a–e), indicating smooth and complete conversion from c-CoO to hollow c-Co nanoparallelopipeds.

Evolution of c-Co hollow nanoparallelepipeds with time: (a) TEM image of c-CoO nanoparallelepipeds. (b, c) TEM images of nanoparallelepipeds after heating c-CoO at 290 °C for 1 h (b) and 2 h (c). (d) TEM image of c-Co nanoparallelepipeds prepared after heating c-CoO at 290 °C 2 h and then at 270 °C for 1 h. A HRTEM image of each of the nanoparallelepipeds is shown as an inset. Adapted from Nam et al.48

XRD patterns during conversion. (a) c-CoO nanoparallelepipeds. (b to d) samples after heating c-CoO at 290 °C for 0.5 h (b), 1 h (c) and 2 h (d). (e) c-Co nanoparticles prepared after heating c-CoO at 290 °C for 2 h and further at 270 °C for 1 h. (f) Proposed reaction pathways: the reduction of c-CoO to c-Co by oleylamine with formation of heptadecene and heptadecadiene. Adapted from Nam et al.48
During the transformation of c-CoO to hollow c-Co, CO and NH3, surprisingly, evolve with a notable absence of O2, and heptadecene is a third co-product identified by GC-MS.48 Plausible reaction pathways are proposed in Figure 6f, including coordination of the amino group on the cobalt site and the C–N bond cleavage of oleylamine. Subsequent reaction sequences are β-H elimination in alkoxide, oxidative addition of C–H bonds, reductive elimination of NH3 and heptadecene, and CO deinsertion and dissociation. The surface heptadecenyl species undergoes β-H elimination to produce heptadecadiene. Overall, c-CoO has been reduced to c-Co by oleylamine, which is oxidized to CO, NH3, and heptadecene. In the case of h-CoO nanoparticles, however, a fast reduction of h-CoO has occurred with the formation of irregular shaped c-Co nanoparticles under the present reduction conditions, suggesting that the abrupt evolution of CO and NH3 gases provides stress that leads to the destruction of the original morphology of h-CoO nanoparticles.
The hollow c-Co nanoparallelopiped would be formed by continuous diffusion of oxygen anions out to the surface voids followed by removal as carbon monoxide, as well as by simultaneous electron transfer from the surface Co atoms to the inner Co2+ ions to form surface Co2+ ions and interior Co atoms. The surface Co metal layer formed at the early stage determines the shape of the hollow c-Co nanoparallelopipeds, and the surface voids diffuse into the interior of the nanoparallelopipeds, resulting in spherical voids, which is entirely due to the loss of oxygen anions. This observation on the formation of the hollow c-Co nanoparallelopipeds, importantly, opens a new synthetic methodology for hollow metal nanoparticles.
The hollow c-Co nanoparallelopiped is ferromagnetic and attracted by a magnet and its saturation magnetization (Ms) has a smaller value (~22%) than those in bulk form as a result of the hollow nature and the large surface-to-volume ratio of the c-Co nanoparallelopiped.
Physicochemical properties of CoO: magnetic and electrochemical properties of h-CoO
The hexagonal metal oxides with magnetically-ordered tetrahedral metal ions, such as Co2+ and Mn2+, are expected to provide coupling magnetism and electric polarization,17 which would offer an extra degree of freedom in the design of conventional devices. The temperature-dependent magnetic measurements indicate that the h-CoO nanoparticles exhibit a paramagnetic behavior (Figure 7a), while the c-CoO nanoparticles are antiferromagnetic.20 The most likely cause of these different magnetic properties is that they have a different structural environment: tetrahedral or octahedral coordination of the oxygen anions around the magnetic Co2+ ions. On the other hand, recent investigation on the magnetic property of h-CoO nanoparticles indicated the particle size dependency of the Néels temperature, at which the transition between antiferromagnetic and paramagnetic states occurs.26, 27 Further research is need in order to clarify the exact magnetic structure of the h-CoO phase.

(a) Zero-field-cooled (ZFC) and field-cooled (FC) curves of h-CoO nanocrystals measured at 100 Oe. (b) First discharge graphs of c-CoO and h-CoO nanocrystals. (c) Initial charge−discharge curves and (d) cycling performance of h-CoO based nanostructures. Adapted from Seo et al.,20 Nam et al.23 and Nam et al.44
Cobalt oxides are known to have superior electrochemical features as negative electrode materials in lithium ion batteries.39 The electrochemical properties of h-CoO and c-CoO nanoparticles have been investigated by means of the assembly of Li/CoO half-cells (Figure 7b).23 The first discharge curve of c-CoO exhibits a characteristic feature with a well-defined potential plateau at 0.8 V vs Li+/Li, which is attributed to the reduction of c-CoO to metallic Co.34 The first discharge graph of h-CoO exhibits a potential plateau of 1.2 V (Figure 7b). The potentials of electrode materials during the reaction with lithium are closely related to the Gibbs free energies of their electrochemical reactions. Therefore, the difference of 0.4 V between the potential plateaus of c-CoO and h-CoO is attributed to the apparent disparity in their reaction free energies. The local structure of c-CoO is composed of CoO6 octahedra, whereas that of h-CoO is CoO4 tetrahedra. The crystal field stabilization energy (CFSE) of the octahedral coordination is larger than that of the tetrahedral coordination in weak field d7 (Co2+) oxides, which accounts for the reduction of h-CoO to Co being much easier than that of c-CoO.
The electrochemical properties of h-CoO nanocrystals, such as h-CoO nanopyramids, h-CoO nanorods and Cu-doped h-CoO nanorods, are also investigated using the Li/h-CoO half cells (Figure 7c and d).23, 44 After the first discharge/charge cycle, the capacity curve exhibits high reversibility with a stable capacity of 944 mA h g−1 after 50 cycles for the Cu-doped h-CoO nanorods. Under the present electrochemical conditions, h-CoO nanopyramids have a moderate value of 617 mA h g−1 and h-CoO nanorods exhibit a poor capacity of 256 mA h g−1. The high capacity of the Cu-doped h-CoO nanorods may originate from a unique architecture of bundled nanorods and enhanced stability due to the Cu doping. This would shorten the diffusion length of Li+ ions, stabilize the active domain structures, and reduce the strain by the volume change during electrochemical reactions.
Hexagonal phase of MnO
A missing hexagonal phase of MnO
Manganese oxide is known to have a stable cubic structure, as do other metal monoxides. Numerous preparation methods of c-MnO nanocrystals have been developed, mainly based on thermal decomposition of Mn precursors in hydrocarbon solvents.3 The physicochemical properties of c-MnO have been extensively studied due to its potential applications such as magnetic resonance imaging (MRI) contrast reagents, battery materials and catalysts.49, 50 The h-MnO phase, however, has long been undiscovered. A theoretical study of hypothetical h-MnO has been reported to show a large piezoelectric response with a unique magnetism,51 which has motivated the discovery and investigation of this unprecedented h-MnO.
Recently, Nam et al.29 have successfully prepared h-MnO. The h-MnO nanoparticles were synthesized via thermal decomposition of a Mn precursor, Mn(acac)2, in the presence of carbon spheres. The TEM and HRTEM images demonstrate an urchin-like architecture of h-MnO nanocrystals on the carbon sphere template (Figure 8a and b). The Rietveld refinement using the XRD data is indexed to hexagonal MnO with a space group of P63mc (Figure 8c).29 In the tetrahedral coordination geometry of the Mn atoms with the oxygen atoms, the dihedral angles of O-Mn-O are 110.55° and 108.38°, and the interatomic distances are a short Mn-O bond of 1.963 Å and three longer bonds of 2.0789 Å, indicating an unusually distorted hexagonal wurtzite lattice (Figure 8c). The energy-dispersive X-ray spectroscopy (EDS) estimated an average atomic ratio of 48.7:51.3 (Mn/O), which was within an acceptable range of the 1:1 atomic composition of h-MnO.29 Rapid conversion of h-MnO to c-MnO occurred during the annealing process, which indicates a distinct rearrangement rate between the manganese cations and the oxygen anions.30 Rearrangement of the oxygen lattice occurs rapidly to form the ccp array under the thermal reaction condition, resulting in the conversion of h-MnO to c-MnO. Since c-MnO has been solely detected in the sample after annealing, it is reasonable to conclude that the hexagonal-to-cubic phase transformation is irreversible.

(a) Bright-field TEM image of h-MnO on the carbon sphere template. (b) HRTEM image of h-MnO. (c) Rietveld refinement patterns of h-MnO using X-ray powder diffraction. Adapted from Nam et al.29
Phase controlled syntheses of h-MnO, c-MnO and tetragonal-Mn3O4
Manganese oxides have a wide variety of stoichiometric components, including MnO, Mn3O4, Mn2O3 and MnO2, with variable oxidation states of manganese atoms.52, 53 To understand the formation of h-MnO, the correlation between crystallographic structures and formation kinetics of MnO and Mn3O4 phases has been explored.30 Thermal decomposition of Mn(acac)2 in benzylamine with carbon spheres under an inert atmosphere yields two distinct MnO phases, via either kinetic or thermodynamic reaction control, as similarly observed in the CoO system (Scheme 1). Rapid heating of the reaction mixture at refluxing temperature produces h-MnO (Reaction A), whereas prolonged heating at a relatively low temperature yields c-MnO (Reaction B), as shown in Scheme 3. The existence of the carbon template may reduce the surface energy and enhance the stability of MnO, leading to the formation of h-MnO. As a control experiment, when a reaction mixture of Mn(acac)2 in benzylamine is heated without carbon spheres, c-MnO nanoparticles are formed along with a small amount of tetragonal-Mn3O4 (t-Mn3O4) phase.30 Furthermore, when a similar reaction is carried out in benzyl alcohol or a mixture of benzylamine and benzyl ether, pure t-Mn3O4 (space group: I41/amd) is obtained (Reaction C).
Thermal decomposition of a Mn(acac)2 precursor in the presence of water typically yields a c-MnO phase (Reaction D of Scheme 3).3 Similar to the formation mechanism of c-CoO (Pathway C of Scheme 2), the decomposition of the manganese hydroxide intermediate is decelerated, and the reaction enters into the regime of thermodynamic control and yields c-MnO nanocrystals. The presence of water, presumably, prohibits further oxidation of the MnO phase to Mn3O4. Water is involved in the decomposition of the acetylacetonate ligands into carboxylates, which is tightly bound to the metal center through chelation and stabilizes metal complexes.
Physicochemical properties of h-MnO
Seo et al.3 have intensively studied the magnetic properties of c-MnO nanocrystals and have reported that c-MnO nanocrystals exhibit a weak ferromagnetic behavior at low temperatures, although bulk c-MnO presents an antiferromagnetic behavior. The observed weak ferromagnetism is due to the presence of noncompensated surface spins on the antiferromagnetic core of the c-MnO nanoparticle. Moreover, the blocking temperature (TB) of the c-MnO nanoparticle is inversely proportional to the particle size,3 which can be ascribed to the higher fraction of noncompensated surface spins for a smaller MnO particle with a higher surface-to-volume ratio.
The h-MnO nanocrystals are paramagnetic (Figure 9a).29 There are no observable magnetic transitions, either by temperature-dependent or field-dependent magnetization, presumably, due to the nanoscale architecture of h-MnO. The energy differences in distinct magnetic orderings become extremely small when the sample size is on the nanoscale, and these differences are related to the strong demagnetization effect due to the large surface area. In order to identify the magnetic ordering and determine the stable MnO phases, density functional calculations are carried out.29 The energy-volume analysis indicates that h-MnO is stabilized in a larger volume compared to c-MnO (Figure 9b), and the stable magnetic structure has an antiferromagnetic ordering. Kan et al. recently found that a h-MnO thin film could transform into a graphitic structure using a density functional calculation.31 The antiferromagnetic ordering of this graphene-like MnO can be easily switched to half-metallic ferromagnetism by a slight doping, suggesting that h-MnO is a promising material for two-dimensional spintronics.

(a) Zero-field-cooled (ZFC) and field-cooled (FC) curves of h-MnO nanocrystals measured at 100 Oe. (b) Total energy vs volume of ferromagnetic ordering (FM) and the antiferromagnetic ordering (AFM) for cubic rock-salt (RS) and hexagonal wurtzite MnO (WZ) structures by density functional theory calculations. Adapted from Nam et al.29
Hexagonal phase of ZnO
Preparation and growth mechanism of h-ZnO
Under ambient conditions, the thermodynamically stable phase of zinc oxide is h-ZnO, which has a hexagonal unit cell with two lattice parameters a and c in the ratio of 1.633 (in an ideal structure), and belongs to the space group of P63mc.33 Zinc blende ZnO is known as a metastable phase and can be stabilized by heteroepitaxial growth on typical substrates such as ZnS and GaAs/ZnS.54 The c-ZnO structure is obtained only at high pressures (around 10 GPa).33, 54 Numerous synthetic techniques for h-ZnO have been developed, including chemical vapor deposition, molecular beam epitaxy, sputtering, electrospinning and wet chemical methods.55, 56, 57 Although many review papers that cover various aspects of nano-structured ZnO materials previously appeared,56, 57 herein we specifically focus on the preparation of h-ZnO using wet chemical methods, and discuss on the crystal growth and phase transition of h-ZnO.
Generally, h-ZnO is formed by the hydrolysis of Zn salts in a basic solution. Depending on the chemical environment, Zn2+ ions exist as a series of intermediates such as Zn(OH)2 and [Zn(OH)4]2-, which ultimately generate h-ZnO crystals upon dehydration.57, 58, 59 In terms of crystal morphology, chemical reactions are usually regarded to be in a reversible equilibrium, and the driving force of the direction of forming the specific crystal morphology is given by minimizing a Gibbs free energy in the entire reaction system.57 Under these thermodynamic equilibrium conditions, low surface energy facets tend to be exposed outward, while high surface energy facets are not. Because polar {001} facets have the highest surface energy with alternating Zn2+-terminated and O2--terminated surfaces, the h-ZnO crystals grow along the [001] direction (c axis), and nonpolar {110} and {100} facets remain on the surface.42 This explains how the h-ZnO is grown to yield the elongated morphology, such as nanorods or nanowires (Figure 10), in accordance with the formation of h-CoO nanostructure (Figure 3d).

TEM images of ZnO nanowires synthesized in solvents having different polarities: (a) in methanol, (b) in ethanol, and (c) in 1-butanol. (d) Schematic illustration of [001] directed nanowires. Adapted from Xu and Wang.57
Since the growth of h-ZnO is highly sensitive to reaction solvents, its morphology could be controlled by crystal-solvent interfacial interactions.58 For instance, the aspect ratio of h-ZnO nanowires was readily tuned by varying the solvent polarity (Figure 10). Polar solvent molecules have strong interactions with the polar surface of h-ZnO, which suppresses the precursor molecules settling down onto the polar surface. The aspect ratio of the h-ZnO nanowires increased when using less polar solvents in order as methanol<ethanol<1-butanol. In addition, the solvent mixture with nonpolar hexane provided ultrathin h-ZnO nanowires with a diameter of 2 nm with a simple acetate precursor.60
The addition of capping agents can modify the growth direction of h-ZnO nanostructures.57, 61 Amines are adsorbed on the side faces of ZnO and enhance a vertical growth along the c axis. The protonated amines over a wide range of pH are strongly adsorbed on the negatively charged surfaces, resulting in hindering a lateral growth of the nanowires.62 In contrast, citrate ions, which have highly negative charges, cap the basal planes of ZnO and promote the lateral growth.63 Experimental and theoretical results reveal that citrate ions are strongly and specifically bound to the Zn2+ ions on the (001) surface, which inhibit the growth along [001] and promote the growth along <100> or <110> direction. As a result, flat hexagonal nanoplates are the major product instead of nanowires.63, 64 The actual processes, however, could be much more complicated, and thus further studies are needed in order to address the exact growth mechanism of ZnO.
Pressure-induced phase transition of h-ZnO
Since the discovery of c-ZnO at relatively high pressures,33 intensive investigations were carried out on pressure-dependent behaviors of the ZnO structure. When the applied pressure increases, a phase transition occurs from h-ZnO to c-ZnO with a large decrease in volume (~17%).33, 34 Due to the strong tetrahedral preference of Zn cations, the phase transition behavior of h-ZnO is distinct from those of h-CoO and h-MnO. The phase change of h-ZnO recovers its original phase after the pressure release, that is, h-ZnO→c-ZnO (pressure)→h-ZnO (release),36, 65 while those of h-CoO and h-MnO are irreversible.
Recently, Baranove et al.37 reported that the c-ZnO can be obtained by the compression of h-ZnO nanopowders. Upon heating at 15 GPa, c-ZnO appears in the diffraction patterns around 600 K, and complete disappearance of h-ZnO reflections is observed at 800 K. The resulting c-ZnO retains its structure even at ambient temperatures. Figure 11a shows a characteristic powder XRD pattern of the cubic phase from recovered samples at an ambient condition (quenched from 7.7 GPa and 800 K). Rietveld refinement has shown that the recovered samples have a single phase, indexed as a cubic rocksalt structure (Fm3m space group, a=4.2813 Å). The c-ZnO persists for at least several months at ambient pressure and temperature. Although bulk samples of nanocrystalline c-ZnO do not show any tendency of phase transition to h-ZnO under normal conditions, intensive grinding of the samples promotes the reverse transition as c-ZnO→h-ZnO. Thermal stability of the resulting c-ZnO nanocrystal has been studied by high temperature synchrotron X-ray diffraction (Figure 11b). The progress of the phase transition has been monitored by mild heating (323~393 K). The formation of h-ZnO has been observed at 333 K (c-ZnO→h-ZnO), and the starting c-ZnO has been completely consumed at 393 K. The nanometer size plays a critical role in the relative stability of the non-native c-ZnO phase. During the pressure-induced phase transition, h-ZnO nanocrystals with a grain size smaller than 45 nm convert directly into c-ZnO, while the use of microcrystalline powders leads to the reconstruction of h-ZnO.35 Micron-sized particles are more likely to reconvert into h-ZnO after the pressure release due to spontaneous intragrain nucleation, which is suppressed in nanoparticles.

(a) Rietveld refinement patterns of the nanocrystalline c-ZnO quenched from 7.7 Gpa and 800 K using X-ray powder diffraction. (b) XRD patterns of nanocrystalline c-ZnO collected in situ upon stepwise heating. Adapted from Baranov et al.37
Physicochemical properties of h-ZnO
The h-ZnO materials have numerous applications such as sensors, lasers, light-emitting diodes, field emission devices, solar cells and nano-piezotronics due to their unique chemical and structural properties.66, 67 Typically, h-ZnO possesses piezoelectric properties originated by its crystal structure, where the Zn2+ cations and O2− anions are tetrahedrally coordinated, and stacked in a layer by layer fashion along the c axis (Figure 12a). The lack of central symmetry on the hexagonal structure results in piezoelectric property, which is vital for mechanic-electric energy conversion in ZnO-based nanogenerators and current modulation in strain sensors. The presence of piezopotential is the fundamental physical basis for the nanogenerators and piezotronics.67 The principle of the nanogenerator is the transient flow of electrons in external load as driven by the piezopotential created by dynamic straining. On the other hand, if the material is a semiconductor, the piezopotential acts as a gate voltage that can tune the transport process of the charge carriers under the driving force of an externally applied voltage. The device fabricated based on this principle is called as the piezotronic device. Transversely deflected ZnO nanowires are normally utilized in nanogenerators for energy harvesting application, while axially strained nanowires are used in piezotronic applications on flexible substrate (Figure 12b).67

Piezopotential in a wurtzite crystal. (a) Atomic model of the wurtzite structured ZnO. (b) The distribution of piezopotential under an axially strained nanowire and a transversely deflected nanowire. Adapted from Wang.67
Semiconductors and magnetic materials are the basis of most of recent technological devices. Materials that share both of semiconducting and magnetic properties, sometimes referred to as dilute magnetic semiconductors (DMS), attracted much interests for a new generation of devices such as spin transistors and spin-polarized light emitting diodes.17 Diamagnetic ZnO can convert to ferromagnetic materials by doping with ferromagnetic elements such as Fe, Ni, Co and Mn.68, 69 Despite some promising results on Mn-doped ZnO,70 it is not clear whether DMS could satisfy the desired high temperature ferromagnetism. The recent experimental results throw doubt on the real origin of magnetism. Some experimental results indicate that the magnetism is originated from the segregation of metallic clusters.71, 72 Other viewpoints suggest that the magnetic properties are not exclusively related to the presence of magnetic ions but strongly determined by defects with different valence states.71 Capping reagents are also one of the potential causes for altering the magnetic property of ZnO nanoparticles and leading to the appearance of room temperature ferromagnetism despite the diamagnetic character of bulk ZnO.73 The h-ZnO nanoparticles were prepared by a sol-gel method and subsequently capped with three different organic molecules (tryoctylphophine, dodecylamine and docecanethiol).73 The EDS analysis was in agreement with the ideal 1:1 composition of ZnO. Figure 13 shows the magnetization curves of h-ZnO nanoparticles, exhibiting diamagnetic characters similar to bulk h-ZnO. However, for thiol-capped h-ZnO nanoparticles, ferromagnetic contribution is clearly observed after subtracting diamagnetic backgrounds (Figure 13c and d). Although the origin of magnetism is still a matter of debate, the magnetic properties of the h-ZnO nanoparticles are assumed to be originated from the two sources, the magnetic moments of the 3d ion impurities behaving as ferromagnetic and an additional magnetic contribution by defects in the nanoparticles.

Magnetization curves loops for ZnO NPs capped with different molecules at (a) 5 K, and (b) 300 K. (c, d) The curves alter subtracting the diamagnetic/paramagnetic background. Adapted from Garcia et al.73
Conclusions and perspectives
This review summarizes recent studies on the syntheses of CoO, MnO and ZnO materials, including an unprecedented, non-native structure and their unique physicochemical properties. Lattice parameters and M-O bond distances of two non-native hexagonal phases (h-CoO and h-MnO) and one new cubic phase (c-ZnO) have been now summarized in Table 1, and h-CoO, h-MnO and c-ZnO have been finally listed in the crystal structures of transition metal monoxides as shown in Figure 1. In the hexagonal phases of CoO, MnO and ZnO, the metal cations sit in tetrahedral sites, resulting in a large reduction in M-O bond distances from those of the cubic phases. The h-CoO and h-MnO materials were successfully prepared via kinetic reaction control during syntheses, although the hexagonal phase is thermodynamically unstable in both materials. The oxidation and reduction behaviors of h-CoO and c-CoO nanocrystals have been fully characterized. The h-CoO nanocrystals are paramagnetic, and exhibit a higher reduction potential than that of the c-CoO phase. The hexagonal phase has been discovered in the MnO system by the kinetic reaction control on the active carbon surface. The h-MnO nanocrystals exhibit a paramagnetic behavior, which was not observed in the c-MnO phase. The carbon template plays a critical role in the formation of h-MnO phase. In the ZnO system, the hexagonal phase of h-ZnO is thermodynamically more stable, and the non-native c-ZnO phase can be obtained by the high-pressure transformation of h-ZnO as the form of nanocrystallites. The h-ZnO nanostructures have been intensively employed to industrial applications such as active materials for nanogenerators and nanopiezotronic devices due to their strong piezoelectricity arose from the intrinsic wurtzite structure.
Since the non-native h-CoO and h-MnO materials have an identical crystal structure with that of the native h-ZnO phase (Scheme 4), structure-related physicochemical properties are naturally expected for the three materials. The piezoelectric constant of h-MnO is calculated as 0.9973 C m−2.29 This value is comparable to that of h-ZnO (0.76–1.18 C m−2),74 which is the highest piezoelectric tensor among semiconductors with tetrahedrally-surrounded cations. This close relationship between the phases and their physicochemical properties could find useful applications of these non-native materials. For example, a problem of p–n doping asymmetry is known to exist in wide band gap semiconductors such as ZnO.75 The relatively small band gap may help to avoid this problem, and allows the materials to have both n-type and p-type conductivities. Zhao et al. proposed a theoretical tuning of p–n conductivity in hexagonal metal monoxides such as h-CoO and h-MnO,32 which have small band gaps applicable for electronic devices. Typically, Fermi levels determined by self-consistent calculations indicate that the h-MnO phase has p-type conductivity under an oxygen-rich condition and conversely exhibits n-type conductivity under an oxygen-poor condition. This excellent control of the p–n conductivity in h-MnO suggests a new promising switching material for electronic devices. The non-native c-ZnO is expected not only to have similar structure-related physicochemical properties as shown in native c-CoO and c-MnO (Scheme 4), but also to exhibit unique physicochemical properties due to the constituent Zn element useful for wide applications in various fields.38, 49, 76, 77 Since ZnO has a strong hexagonal structure preference under ambient conditions, the development of reliable synthetic methods for c-ZnO would open new potential applications such as catalysts, lithium-ion battery, light emitting diode, and solid-state sensors in the near future.
For the preparation of the non-native phases, careful control of kinetic parameters is required during synthetic processes, since the non-native phases have been observed under a narrow range of the reaction conditions. Moreover, the non-native phases are only found in the nanometer range due to the energy required for lattice distortion to the non-native phase being lowered only on the nanometer scale. The surface energy of nanomaterials plays a dominant role in the stabilization of the non-native phases. Consequently, the development of new synthetic strategies together with additional theoretical studies is indispensable to gain further fruitful results in this field. For instance, hexagonal phases of h-NiO and h-FeO are theoretically hypothesized but not observed.78, 79
Further intensive research on designed syntheses and physicochemical properties of the non-native phases of transition metal monoxides would provide an enormous impact on materials chemistry and open an unprecedented avenue in magnetic, catalytic, electronic and spintronic applications of these new materials.

Selective formation of h-CoO and c-CoO via either kinetic or thermodynamic reaction control.

(a) Reaction pathways for the formation of h-CoO nanocrystals (Pathways A and B), and (b) a pathway for the formation of c-CoO nanocrystals (Pathway C) in the presence of water. The products are as follows: benzylamine (1), N-benzylacetamide (2), N-isopropylidenebenzylamine (3) and 4-benzylamino-3-penten-2-one (4). Adapted from Shim et al.39

Phase-selective syntheses of h-MnO and c-MnO via kinetic (Reaction A) and thermodynamic reaction control (Reaction B) on the carbon sphere template, and syntheses of c-MnO and t-Mn3O4 without (Reaction C) or with H2O (Reaction D) during thermal decomposition.

Crystal structures of the MnO, CoO and ZnO materials.
References
Rdddy, M. V., Subba Rao, G. V. & Chowdari, B. V. R. Metal oxides and oxysalts as anode materials for li ion batteries. Chem. Rev. 113, 5364–5457 (2013).
Jun, Y.-W., Choi, J.-S. & Cheon, J. Shape control of semiconductor and metal oxide nanocrystals through nonhydrolytic colloidal routes. Angew. Chem. Int. Ed. 45, 3414–3439 (2006).
Seo, W. S., Jo, H. H., Lee, K., Kim, B., Oh, S. J. & Park, J. T. Size-dependent magnetic properties of colloidal Mn3O4 and MnO nanoparticles. Angew. Chem. Int. Ed. 116, 1135–1137 (2004).
Tao, J., Luttrell, T. & Batzill, M. A two-dimensional phase of TiO2 with a reduced bandgap. Nat. Chem 3, 296–300 (2011).
Moreo, A., Yunoki, S. & Dagotto, E. Phase separation scenario for manganese oxides and related materials. Science 283, 2034–2040 (1999).
Kung, H. H. Transition Metal Oxides: Surface Chemistry and Catalysis Vol. 45; (Elsevier: NY, USA, 1989).
Miessler, G. L., Fischer, P. J. & Tarr, D. A. Inorganic Chemistry 5th edn (Pearson Education: NJ, USA, 2014).
Roth, W. L. Magnetic structure of MnO, FeO, CoO, and NiO. Phys. Rev 110, 1333–1341 (1958).
Zhang, D., Wong, A. B., Yu, Y., Brittman, S., Sun, J., Fu, A., Beberwyck, B., Alivisatos, A. P. & Yang, P. Phase-selective cation-exchange chemistry in sulfide nanowire systems. J. Am. Chem. Soc. 136, 17430–17433 (2014).
Murray, C. B., Norris, D. J. & Bawendi, M. G. Synthesis and characterization of nearly monodisperse CdE (E=sulfur, selenium, tellurium) semiconductor nanocrystallites. J. Am. Chem. Soc. 115, 8706–8715 (1993).
Chen, C.-C., Herhold, A. B., Johnson, C. S. & Alivisatos, A. P. Size dependence of structural metastability in semiconductor nanocrystals. Science 276, 398–401 (1997).
Dinega, D. P. & Bawendi, M. G. A solution-phase chemical approach to a new crystal structure of cobalt. Angew. Chem. Int. Ed. 38, 1788–1791 (1999).
Sakurai, S., Namai, A., Hashimoto, K. & Ohkoshi, S.-I. First observation of phase transformation of all four Fe2O3 phases (γ→ɛ→β→α-Phase). J. Am. Chem. Soc. 131, 18299–18303 (2009).
Sun, Y., Mayers, B. & Xia, Y. Metal nanostructures with hollow interiors. Adv. Mater. 15, 641–646 (2003).
Sines, I. T., Misra, R., Schiffer, P. & Schaak, R. E. Colloidal synthesis of non-equilibrium wurtzite-type MnSe. Angew. Chem. Int. Ed. 49, 4638–4640 (2010).
Yang, X., Wang, Y., Sui, Y., Huang, X., Cui, T., Wang, C., Liu, B., Zou, G. & Zou, B. Morphology-controlled synthesis of anisotropic wurtzite MnSe nanocrystals: optical and magnetic properties. CrystEngComm 14, 6916–6920 (2012).
Ohno, H. Making nonmagnetic semiconductors ferromagnetic. Science 281, 951–956 (1998).
Han, H. Elastic, piezoelectric and acoustic properties of wurzite MnO from density functional calculation. Commun. Theor. Phys. 59, 751–755 (2013).
Redman, M. J. & Steward, E. G. Cobaltous oxide with the zinc blende/wurtzite-type crystal structure. Nature 193, 867 (1962).
Seo, W. S., Shim, J. H., Oh, S. J., Lee, E. K., Hur, N. H. & Park, J. T. Phase- and size-controlled synthesis of hexagonal and cubic CoO nanocrystals. J. Am. Chem. Soc. 127, 6188–6189 (2005).
Risbud, A. S., Snedeker, L. P., Elcombe, M. M., Cheetham, A. K. & Seshadri, R. Wurtzite CoO. Chem. Mater. 17, 834–838 (2005).
An, K., Lee, N., Park, J., Kim, S. C., Hwang, Y., Park, J.-G., Kim, J.-Y., Park, J.-H., Han, M. J., Yu, J. & Hyeon, T. Synthesis, characterization, and self-assembly of pencil-shaped CoO nanorods. J. Am. Chem. Soc. 128, 9753–9760 (2006).
Nam, K. M., Shim, J. H., Han, D.-W., Kwon, H. S., Li, Y., Song, H., Seo, W. S. & Park, J. T. Syntheses and characterization of wurtzite CoO, rocksalt CoO, and spinel Co3O4 nanocrystals: their interconversion and tuning of phase and morphology. Chem. Mater. 22, 4446–4454 (2010).
Buck, M. B., Biacchi, A. J. & Schaak, R. E. Insights into the thermal decomposition of Co (II) oleate for the shape-controlled synthesis of wurtzite-type CoO nanocrystals. Chem. Mater. 26, 1492–1499 (2014).
Walsh, S. R., Rusakova, I. & Whitmire, K. H. Rock salt vs. wurtzite phases of Co1-xMnxO: control of crystal lattice and morphology at the nanoscale. CrystEngComm 15, 775–784 (2013).
He, X., Zhong, W., Yan, S., Liu, C., Shi, H., Au, C.-T. & Du, Y. Transition temperature of wurtzite CoO nanocrystals as revealed in comprehensive magnetic characterization. J. Phys. Chem. C. 118, 13898–13903 (2014).
He, X. M., Zhong, W. & Du, Y. W. Hexagonal CoO nanoparticles as studied by electron spin resonance. J. Appl. Phys. 117, 043905 (2015).
Lu, A., Chen, Y., Zeng, D., Li, M., Xie, Q., Zhang, X. & Peng, D.-L. Shape-related optical and catalytic properties of wurtzite-type CoO nanoplates and nanorods. Nanotechnology 25, 035707 (2014).
Nam, K. M., Kim, Y.-I., Jo, Y., Lee, S. M., Kim, B. G., Choi, R., Choi, S.-I., Song, H. & Park, J. T. New crystal structure: synthesis and characterization of hexagonal wurtzite MnO. J. Am. Chem. Soc. 134, 8392–8395 (2012).
Nam, K. M. & Park, J. T. Chemical approach to a new crystal structure: phase control of manganese oxide on a carbon sphere template. Chem. Asian J. 9, 3525–3532 (2014).
Kan, E., Li, M., Hu, S., Xiao, C., Xiang, H. & Deng, K. Two-dimensional hexagonal transition-metal oxide for spintronics. J. Phys. Chem. Lett. 4, 1120–1125 (2013).
Wu, J.-M., Chen, L.-J., Yang, X.-B. & Zhao, Y.-J. Tuning p/n conductivity in wurtzite transition metal monoxide: Role of native defects in CoO and MnO. Phys. Lett. A 378, 2635–2639 (2014).
Bates, C. H., White, W. B. & Roy, R. New high-pressure polymorph of zinc oxide. Science 137, 993 (1962).
Kusaba, K., Syono, Y. & Kikegawa, T. Phase transition of ZnO under high pressure and temperature. Proc, Jpn Acad. 75(B), 1–6 (1999).
Solozhenko, V. L., Kurakevych, O. O., Sokolov, P. S. & Baranov, A. N. Kinetics of the wurtzite-to-rock-salt phase transformation in ZnO at high pressure. J. Phys. Chem. A 115, 4354–4358 (2011).
Dong, Z., Zhuravlev, K. K., Morin, S. A., Li, L., Jin, S. & Song, Y. Pressure-induced structural transformations of ZnO nanowires probed by X-ray diffraction. J. Phys. Chem. C 116, 2102–2107 (2012).
Baranov, A. N., Sokolov, P. S., Tafeenko, V. A., Lathe, C., Zubavichus, Y. V., Veligzhanin, A. A., Chukichev, M. V. & Solozhenko, V. L. Nanocrystllinity as a route to metastable phases: rock salt ZnO. Chem. Mater. 25, 1775–1782 (2013).
Poizot, P., Laruelle, S., Grugeon, S., Dupont, L. & Tarascon, J.-M. Nano-sized transition-metal oxides as negative-electrode materials for lithium-iion batteries. Nature 407, 496–499 (2000).
Shim, J. H., Nam, K. M., Seo, W. S., Song, H. & Park, J. T. The role of water for the phase-selective preparation of hexagonal and cubic cobalt oxide nanoparticles. Chem. Asian J. 6, 1575–1581 (2011).
Pinna, N., Garnweitner, G., Antonietti, M. & Niederberger, M. A. General nonaqueous route to binary metal oxide nanocrystals involving a C-C bond cleavage. J. Am. Chem. Soc. 127, 5608–5612 (2005).
Connolly, J. A., Kim, J. H., Banaszczyk, M., Drouin, M. & Chin, J. Factors affecting chelation of carboxylates to cis-diaqua Co(III) complexed-implications on the reactivity of the metal-complexes for hydrolyzing esters, amides, nitriles, and phosphates. Inorg. Chem. 34, 1094–1099 (1995).
Huang, M. H., Mao, S., Feick, H., Yan, H., Wu, Y., Kind, H., Weber, E., Russo, R. & Yang, P. Room-temperature ultraviolet nanowire nanolasers. Science 292, 1897–1899 (2001).
Kong, X. Y., Ding, Y., Yang, R. & Wang, Z. L. Single-crystal nanorings formed by epitaxial self-coiling of polar nanobelts. Science 303, 1348–1351 (2004).
Nam, K. M., Choi, Y. C., Jung, S. C., Kim, Y.-I., Jo, M. R., Park, S. H., Kang, Y.-M., Han, Y.-K. & Park, J. T. [100] directed Cu-doped h-CoO nanorods: elucidation of the growth mechanism and application to lithium-ion batteries. Nanoscale 4, 473–477 (2012).
Tolbert, S. H. & Alivisatos, A. P. Size dependence of a first order solid-solid phase transition: the wurtzite to rock salt transformation in CdSe nanocrystals. Science 265, 373–376 (1994).
Liu, J. F., Yin, S., Wu, H. P., Zeng, Y. W., Hu, X. R., Wang, Y. W., Lv, G. L. & Jiang, J. Z. Wurtzite-to-rocksalt structural transformation in nanocrystalline CoO. J. Phys. Chem. B 110, 21588–21592 (2007).
Liu, J. F., He, Y., Chen, W., Zhang, G. Q., Zeng, Y. W., Kikegawa, T. & Jiang, J. Z. Bulk modulus and structural phase transition of wurtzite CoO nanocrystals. J. Phys. Chem. C 111, 2–5 (2006).
Nam, K. M., Shim, J. H., Ki, H., Choi, S.-I., Lee, G., Jang, J. K., Jo, Y., Jung, M.-H., Song, H. & Park, J. T. Single-crystalline hollow face-centered-cubic nanoparticles from solid face-centered-cubic oxide nanoparticles. Angew. Chem. Int. Ed. 48, 9504–9508 (2008).
Na, H. B., Lee, J. H., An, K., Park, Y. I., Park, M., Lee, I. S., Nam, D.-N., Kim, S. T., Kim, S.-H., Kim, S.-W., Lim, K.-H., Kim, K.-S., Kim, S.-O. & Hyeon, T. Development of a T1 contrast agent for magnetic resonance imaging using mno nanoparticles. Angew. Chem. Int. Ed. 46, 5397–5401 (2007).
Sangaraju, S. & Gedanken, A. MnO oxtahedral nanocrystals and MnO@C core-shell composites: synthesis, characterization, and electrocatalytic properties. J. Phys. Chem. B 110, 24486–24491 (2006).
Gopal, P., Spaldin, N. A. & Waghmare, U. V. First-principles study of wurtzite-structure MnO. Phys. Rev. B 70, 205104 (2004).
Rusakova, I., Ould-Ely, T., Hofmann, C., Prieto-Centurion, D., Levin, C. S., Halas, N. J., Luettge, A. & Whitmire, K. H. Nanoparticle shape conservation in the conversion of MnO nanocrosses into Mn3O4 . Chem. Mater. 19, 1369–1375 (2007).
Robinson, D. M., Go, Y. B., Mui, M., Gardner, G., Zhang, Z., Mastrogiovanni, D., Garfunkel, E., Li, J., Greenblatt, M. & Dismukes, G. C. Photochemical water oxidation by crystalline polymorphs of manganese oxides: structural requirements for catalysis. J. Am. Chem. Soc. 135, 3494–3501 (2013).
Ozgur, U., Alivov, Y. I., Liu, C., Teke, A., Reshchikov, M. A., Dogan, S., Avrutin, V., Cho, S.-J. & Morkoc, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 98, 041301 (2005).
Wang, Z. L. ZnO nanowire and nanobelt platform for nanotechnology. Mater. Sci. Eng. R 64, 33–71 (2009).
Li, J., Ma, S., Liu, X., Zhou, Z. & Sun, C. Q. ZnO meso-mechano-thermo physical chemistry. Chem. Rev. 112, 2833–2852 (2012).
Xu, S. & Wang, Z. L. One-dimensional ZnO nanostructures: solution growth and functional properties. Nano. Res 4, 1013–1098 (2011).
Cheng, B. & Samulski, E. T. Hydrothermal synthesis of one-dimensional ZnO nanostructures with different aspect ratios. Chem. Commun. 40, 986–987 (2004).
Liu, B. & Zeng, H. C. Hydrothermal synthesis of ZnO nanorods in the diameter regime of 50 nm. J. Am. Chem. Soc. 125, 4430–4441 (2003).
Yin, M., Gu, Y., Kuskovsky, I. L., Andelman, T., Zhu, Y., Neumark, G. F. & O’Brien, S. Zinc oxide quantum rods. J. Am. Chem. Soc. 125, 6206–6207 (2003).
Zhang, H., Yang, D. R., Li, D. S., Ma, X. Y., Li, S. Z. & Que, D. L. Controllable growth of ZnO microcrystals by a capping-molecule-assisted hydrothermal process. Cryst. Growth Des. 5, 547–550 (2005).
Wu, W. B., Hu, G. D., Cui, S. G., Zhou, Y. & Wu, H. T. Epitaxy of vertical ZnO nanorod arrays on highly (001)-oriented ZnO seed monolayer by a hydrothermal route. Cryst. Growth Des. 8, 4014–4020 (2008).
Tian, Z. R., Voigt, J. A., Liu, J., Mckenzie, B. & Mcdermott, M. J. Biomimetic arrays of oriented helical ZnO nanorods and columns. J. Am. Chem. Soc. 124, 12954–12955 (2002).
Wang, Z. L., Yang, R., Zhou, J., Qin, Y., Xu, C., Hu, Y. & Xu, S. Lateral nanowire/nanobelt based nanogenerators, piezotronics and piezo-phototronics. Mater. Sci. Eng. R 70, 320–329 (2010).
Desgreniers, S. High-density phases of ZnO: structural and compressive parameters. Phy. Rev. B 58, 14102–14105 (1998).
Wang, Z. L. Zinc oxide nanostructures: growth, properties and applications. J. Phys.: Condens. Mater 16, R829–R858 (2004).
Wang, Z. L. Piezopotential gated nanowire devices: piezotronics and piezo-phototronics. Nano Today 5, 540–552 (2010).
Ueda, K., Tabata, H. & Kawai, T. Magnetic and electric properties of transition-metal-doped ZnO films. Appl. Phys. Lett. 79, 988–990 (2001).
Rubi, D., Fontcuberta, J., Calleja, A., Aragones, L.l., Capdevila, X. G. & Segarra, M. Reversible ferromagnetic switching in ZnO:(Co,Mn) powders. Phys. Rev. B 75, 155322 (2007).
Sharma, P., Gupta, A., Rao, K. V., Owens, F. J., Sharma, R., Ahuja, R., Guillen, M. O., Johansson, B. & Gehring, G. A. Ferromagnetism above room temperature in bulk and transparent thin films of Mn-doped ZnO. Nat. Mater. 2, 673–67 (2003).
Pan, S., Song, C., Liu, X. J., Yang, Y. C. & Zeng, F. Ferromagnetism and possible application in spintronics of transition-metal-doped ZnO films. Mater. Sci. Eng. R 62, 1–35 (2008).
Garcia, M. A., Ruiz-Gonzalex, M. L., Quesada, A., Costa-Kramer, J. L., Fernandez, J. F., Khatib, S. J., Wennberg, A., Caballero, A. C., Martin-Gonzalez, M. S., Villegas, M., Briones, F., Gonzalez-Calbet, J. M. & Hernando, A. Interface double-exchange ferromagnetism in the Mn-Zn-O system: new class of biphase magnetism. Phys. Rev. Lett. 94, 217206 (2005).
Garcia, M. A., Merino, J. M., Pinel, E. F., Quesada, A., Venta, J. D. L., Ruiz Gonzalez, M. L., Castro, G. R., Crespo, P., Llopis, J., Gonzalez-Calbet, J. M. & Hernando, A. Magnetic properties of ZnO nanoparticles. Nano. Lett. 7, 1489–1494 (2007).
Corso, A. D., Posternak, M., Resta, R. & Baldereschi, A. Ab initio study of piezoelectricity and spontaneous polarization in ZnO. Phys. Rev. B 50, 10715–10721 (1994).
Isberg, J., Hammersberg, J., Johansson, E., Wikstrom, T., Twitchen, D. J., Whitehead, A. J., Coe, S. E. & Scarsbrook, G. A. High carrier mobility in single-crystal plasma-deposited diamond. Science 297, 1670–1672 (2002).
Liao, L., Zhang, Q., Su, Z., Zhao, Z., Wang, Y., Li, Y., Lu, X., Wei, D., Feng, G., Yu, Q., Cai, X., Zhao, J., Ren, Z., Fang, H., Robles-hernandez, F., Baldelli, S. & Bao, J. Efficient solar water-splitting using a nanocrystalline CoO photocatalyst. Nat. Nanotech. 9, 69–73 (2014).
Jin, K., Chu, A., Park, J., Jeong, D., Jerng, S. E., Sim, U., Jeong, H.-Y., Lee, C. W., Park, Y.-S., Yang, K. D., Pradhan, G. K., Kim, D., Sung, N.-E., Kim, S. H. & Nam, K. T. Partially oxidized sub-10 nm MnO nanocrystals with high activity for water oxidation catalysis. Sci. Rep 5, 10279 (2015).
Xiao, J., Cuc, A., Pokhrel, S., Schowalter, M., Parlapalli, S., Rosenauer, A., Frauenheim, T., Madler, L., Pettersson, L. G. M. & Heine, T. Evidence for Fe+2 in wurtzite coordination: iron doping stabilizes ZnO nanoparticles. Small 7, 2879–2886 (2011).
Wu, J., Peng, R. Q., W., G., Liu, L. & Feng, Y. P. Wurtzite NiO: a potential half-metal for wide gap semiconductors. Appl. Phys. Lett. 89, 082504 (2006).
Djerdj, I., Arcon, D., Jaglicic, Z. & Niederberger, M. Nonaqueous synthesis of manganese oxide nanoparticles, structural characterization, and magnetic properties. J. Phys. Chem. C 111, 3614–3623 (2007).
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2015R1C1A1A02037373). WSS thanks for the support of the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korea Government (NRF-2014R1A1A2056619). HS acknowledges the National Research Foundation of Korea (NRF) funded by the Korea Government (MSIP) (NRF-2015R1A2A2A01004196).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Nam, K., Seo, W., Song, H. et al. Non-native transition metal monoxide nanostructures: unique physicochemical properties and phase transformations of CoO, MnO and ZnO. NPG Asia Mater 9, e364 (2017). https://doi.org/10.1038/am.2017.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.38
This article is cited by
-
Piezoelectric properties of zinc oxide/iron oxide filled polyvinylidene fluoride nanocomposite fibers
Journal of Materials Science: Materials in Electronics (2021)
-
Structural, photoluminescence, antibacterial and biocompatibility features of zinc incorporated hydroxyapatite nanocomposites
Journal of Materials Science: Materials in Electronics (2021)
-
Interactions of (MY)6 (M = Zn, Cd; Y = O, S, Se) quantum dots with N-bases
Structural Chemistry (2019)
