
Featured
-
-
Article
| Open Access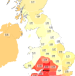 Apparent latent structure within the UK Biobank sample has implications for epidemiological analysis
Apparent latent structure within the UK Biobank sample has implications for epidemiological analysis
Population structure can bias the results of genetic and epidemiological analysis. Here, Haworth et al. report that fine-scale structure is detectable in apparently homogeneous samples such as ALSPAC when measured very precisely, and remains detectable in UK Biobank despite conventional approaches to account for it.
- Simon Haworth
- , Ruth Mitchell
- & Nicholas J. Timpson
-
Article
| Open Access Approximate Bayesian computation with deep learning supports a third archaic introgression in Asia and Oceania
Approximate Bayesian computation with deep learning supports a third archaic introgression in Asia and Oceania
Introgression of Neanderthals and Denisovans left genomic signals in anatomically modern human after Out-of-Africa event. Here, the authors identify a third archaic introgression common to all Asian and Oceanian human populations by applying an approximate Bayesian computation with a Deep Learning framework.
- Mayukh Mondal
- , Jaume Bertranpetit
- & Oscar Lao
-
Article
| Open Access Supplementary stocking selects for domesticated genotypes
Supplementary stocking selects for domesticated genotypes
Stocking of hatchery produced fish is widely used to supplement wild fish populations. Here, the authors show that supplementary stocking can unintentionally favour introgressed individuals with domestic genotypes and compromise the fitness of a wild population of Atlantic salmon.
- Ingerid J. Hagen
- , Arne J. Jensen
- & Sten Karlsson
-
Article
| Open Access Emergence of a floral colour polymorphism by pollinator-mediated overdominance
Emergence of a floral colour polymorphism by pollinator-mediated overdominance
Examples of overdominance are usually explained by deleterious effects in homozygotes. Here, Kellenberger et al. describe a case of overdominance in the floral color of the Alpine orchid Gymnadenia rhellicani apparently maintained by pollinator preferences without deleterious effects in homozygotes.
- Roman T. Kellenberger
- , Kelsey J. R. P. Byers
- & Philipp M. Schlüter
-
Article
| Open Access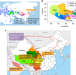 Origin and evolution of qingke barley in Tibet
Origin and evolution of qingke barley in Tibet
The origin of Tibetan barley (qingke) has been a controversial issue for many years. Here, the authors conduct population genomics study to support that qingke is derived from eastern domesticated barley instead of Tibetan wild barley and suggest southern Tibetan Plateau as its introduction route.
- Xingquan Zeng
- , Yu Guo
- & Nyima Tashi
-
Article
| Open Access Dissection of genetic variation and evidence for pleiotropy in male pattern baldness
Dissection of genetic variation and evidence for pleiotropy in male pattern baldness
Male pattern baldness (MPB) is a polygenic trait that affects the majority of European men. Here, Yap et al. estimate heritability, partitioned by autosomes and the X-chromosome, of MPB in the UK Biobank cohort, perform GWAS for MPB and find genetic correlation with other sex-specific traits.
- Chloe X. Yap
- , Julia Sidorenko
- & Peter M. Visscher
-
Article
| Open Access Latin Americans show wide-spread Converso ancestry and imprint of local Native ancestry on physical appearance
Latin Americans show wide-spread Converso ancestry and imprint of local Native ancestry on physical appearance
Latin Americans trace their ancestry to the admixture of Native Americans, Europeans and Sub-Saharan Africans. Here, the authors develop a novel haplotype-based approach and analyse over 6,500 Latin Americans to infer the geographically-detailed genetic structure of this population.
- Juan-Camilo Chacón-Duque
- , Kaustubh Adhikari
- & Andrés Ruiz-Linares
-
Article
| Open Access Genomic insights into multidrug-resistance, mating and virulence in Candida auris and related emerging species
Genomic insights into multidrug-resistance, mating and virulence in Candida auris and related emerging species
Candida auris is an emergent fungal pathogen that is resistant to multiple antifungals. Here, Muñoz et al. analyse genomic sequences for isolates from each of the four major C. auris clades and for three related species, and identify genetic features associated with virulence, antifungal resistance and mating.
- José F. Muñoz
- , Lalitha Gade
- & Christina A. Cuomo
-
Article
| Open Access Northern forest tree populations are physiologically maladapted to drought
Northern forest tree populations are physiologically maladapted to drought
Northern tree populations may not benefit under climate change, with implications for assisted migration and range expansion. Here, Isaac-Renton et al. show that leading-edge lodgepole pine populations have fewer characteristics of drought-tolerance, so may not adapt to tolerate drier conditions.
- Miriam Isaac-Renton
- , David Montwé
- & Kerstin Treydte
-
Article
| Open Access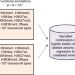 A semi-supervised approach for predicting cell-type specific functional consequences of non-coding variation using MPRAs
A semi-supervised approach for predicting cell-type specific functional consequences of non-coding variation using MPRAs
Predicting the functional consequences of non-coding genetic variants is a challenge. Here, He et al. present GenoNet, a semi-supervised method that combines information from experimentally confirmed regulatory variants with cell type- and tissue specific annotation for function prediction.
- Zihuai He
- , Linxi Liu
- & Iuliana Ionita-Laza
-
Article
| Open Access Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe
Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe
Populations from North-eastern Europe, in particular those speaking Uralic languages, carry additional ancestry in similarity with modern East Asian populations. Here, the authors analyse ancient genomic data from 11 individuals from Finland and Northwest Russia, and identify genomic signals of migrations from Siberia that began at least 3500 years ago.
- Thiseas C. Lamnidis
- , Kerttu Majander
- & Stephan Schiffels
-
Article
| Open Access Genetic variation in PTPN1 contributes to metabolic adaptation to high-altitude hypoxia in Tibetan migratory locusts
Genetic variation in PTPN1 contributes to metabolic adaptation to high-altitude hypoxia in Tibetan migratory locusts
Vertebrate adaptation to high-altitude life has been extensively investigated, while invertebrates are less well-studied. Here, the authors find signals of adaptive evolution in genomes of migratory locusts from the Tibetan Plateau, and implicate a PTPN1 coding mutation in their hypoxia response.
- Ding Ding
- , Guangjian Liu
- & Le Kang
-
Article
| Open Access Demographic histories and genetic diversity across pinnipeds are shaped by human exploitation, ecology and life-history
Demographic histories and genetic diversity across pinnipeds are shaped by human exploitation, ecology and life-history
Historical hunting has caused documented declines in pinnipeds, but the extent to which hunting caused genetic bottlenecks among species was unknown. Here, the authors show evidence of severe bottlenecks in several pinniped species, particularly those that breed on land.
- M. A. Stoffel
- , E. Humble
- & J. I. Hoffman
-
Article
| Open Access Cohort-wide deep whole genome sequencing and the allelic architecture of complex traits
Cohort-wide deep whole genome sequencing and the allelic architecture of complex traits
Rare genetic variants can contribute to complex traits but this contribution is not well understood. Here, the authors analyse deep whole genome sequencing data across 1457 individuals from an isolated Greek population and find association of rare variant burdens with cardiometabolic traits.
- Arthur Gilly
- , Daniel Suveges
- & Eleftheria Zeggini
-
Article
| Open Access A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease
A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease
Mutations in genes encoding NAPDH oxidase subunits are known to be causative for the primary immunodeficiency chronic granulomatous disease (CGD). Here, the authors identify CYBC1 mutations in patients with CGD and show that CYBC1 is important for formation of the NADPH complex and respiratory burst.
- Gudny A. Arnadottir
- , Gudmundur L. Norddahl
- & Kari Stefansson
-
Correspondence
| Open Access The role of MHC supertypes in promoting trans-species polymorphism remains an open question
The role of MHC supertypes in promoting trans-species polymorphism remains an open question
- Maciej J. Ejsmond
- , Karl P. Phillips
- & Jacek Radwan
-
Article
| Open Access Extremely rare variants reveal patterns of germline mutation rate heterogeneity in humans
Extremely rare variants reveal patterns of germline mutation rate heterogeneity in humans
Germline mutation rate is a critical parameter in the study of genetics and evolution. Here, Carlson et al. infer fine-scale patterns of human mutation rate heterogeneity by analyzing ~36 million singleton variants from 3560 whole-genome sequences.
- Jedidiah Carlson
- , Adam E. Locke
- & Stanley J. Watson
-
Article
| Open Access Understanding 6th-century barbarian social organization and migration through paleogenomics
Understanding 6th-century barbarian social organization and migration through paleogenomics
The Longobards invaded and conquered much of Italy after the fall of the Western Roman Empire. Here, the authors sequence and analyze ancient genomic DNA from 63 samples from two cemeteries associated with the Longobards and identify kinship networks and two distinct genetic and cultural groups in each.
- Carlos Eduardo G. Amorim
- , Stefania Vai
- & Krishna R. Veeramah
-
Article
| Open Access Ancient DNA from Chalcolithic Israel reveals the role of population mixture in cultural transformation
Ancient DNA from Chalcolithic Israel reveals the role of population mixture in cultural transformation
The Late Chalcolithic material culture in the southern Levant has unique attributes that suggest spread of people or culture. Here, the authors use genome-wide ancient DNA data from 22 individuals from a Chalcolithic site and show evidence of complex population movements and turnovers.
- Éadaoin Harney
- , Hila May
- & David Reich
-
Article
| Open Access Early selection of bZIP73 facilitated adaptation of japonica rice to cold climates
Early selection of bZIP73 facilitated adaptation of japonica rice to cold climates
Japonica rice can grow further north than wild or indica rice and is more tolerant of cold climates. Here, the authors show that bZIP73 likely underwent selection in the early phase of rice domestication to facilitate cold tolerance in japonica by modulating ABA and ROS homeostasis.
- Citao Liu
- , Shujun Ou
- & Chengcai Chu
-
Article
| Open Access Genome‐wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease
Genome‐wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease
Genetic variation can influence levels of disease-related plasma proteins and, thus, contribute to the pathogenesis of complex diseases. Here, the authors perform genome-wide QTL analysis for 71 plasma proteins to identify causal proteins for coronary heart disease and provide a molecular QTL browser.
- Chen Yao
- , George Chen
- & Daniel Levy
-
Article
| Open Access A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots
A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots
Clustering methods such as STRUCTURE and ADMIXTURE are widely used in population genetic studies to investigate ancestry. Here, the authors provide a tutorial on how to interpret results of these analyses and a tool to test the goodness of fit of the model.
- Daniel J. Lawson
- , Lucy van Dorp
- & Daniel Falush
-
Article
| Open Access An intercross population study reveals genes associated with body size and plumage color in ducks
An intercross population study reveals genes associated with body size and plumage color in ducks
Ducks, one of the most common domestic fowls, originated from mallards. Here, the authors perform whole-genome sequencing of mallards, indigenous-breed ducks, and Pekin ducks, as well as 1026 ducks from a population generated by wild × domestic crosses to identify selection signals and map variants associated with body size and plumage color.
- Zhengkui Zhou
- , Ming Li
- & Yu Jiang
-
Article
| Open Access Gene expression drives the evolution of dominance
Gene expression drives the evolution of dominance
Dominance is difficult to measure in natural populations as it is confounded with fitness. Here, Huber et al. developed a new approach to co-estimate dominance and selection coefficients, and found that the observed relationship is best fit by a new model of dominance based on gene expression level.
- Christian D. Huber
- , Arun Durvasula
- & Kirk E. Lohmueller
-
Article
| Open Access Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination
Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination
Since the 1980s, hypervirulent clonal-group CG23 serotype K1 Klebsiella pneumoniae has been recognised as a prominent cause of community-acquired liver abscess and other severe infections. Here, the authors investigate the genomic evolutionary history of CG23 and suggest a new reference strain for CG23.
- Margaret M. C. Lam
- , Kelly L. Wyres
- & Kathryn E. Holt
-
Article
| Open Access The origin and adaptive evolution of domesticated populations of yeast from Far East Asia
The origin and adaptive evolution of domesticated populations of yeast from Far East Asia
An understanding of the domestication of the yeast Saccharomyces cerevisiae has important implications for studying its evolution and diversity. Here, the authors show that Far East Asia is likely the center of origin of the domesticated populations of the yeast based on genomic and phenotypic characterization of a large collection of isolates.
- Shou-Fu Duan
- , Pei-Jie Han
- & Feng-Yan Bai
-
Article
| Open Access Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries
Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries
Circulating lipoprotein(a) is an important risk factor for cardiovascular disease and shows variability between different ethnic groups. Here, Zekavat et al. perform whole-genome sequencing in individuals of European and African ancestries and find ancestry-specific genetic determinants for lipoprotein(a) levels.
- Seyedeh M. Zekavat
- , Sanni Ruotsalainen
- & Sebastian Zoellner
-
Article
| Open Access Genomic analysis of a pre-elimination Malaysian Plasmodium vivax population reveals selective pressures and changing transmission dynamics
Genomic analysis of a pre-elimination Malaysian Plasmodium vivax population reveals selective pressures and changing transmission dynamics
Plasmodium vivax incidence in Malaysia has declined markedly over the last decade, despite evidence of chloroquine resistance. Here, Auburn et al. compare population structure of P. vivax in Malaysia to regions with intermediate and high transmission and identify genetic regions under putative selection.
- Sarah Auburn
- , Ernest D. Benavente
- & Ric N. Price
-
Article
| Open Access The origin and remolding of genomic islands of differentiation in the European sea bass
The origin and remolding of genomic islands of differentiation in the European sea bass
The speciation process tends to generate ‘genomic islands’ of increased divergence. Here, the authors use haplotype–resolved whole-genome sequences of European sea bass lineages to infer divergence history and show that linked selection generated genomic islands that resist introgression at secondary contact.
- Maud Duranton
- , François Allal
- & Pierre-Alexandre Gagnaire
-
Article
| Open Access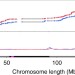 High-resolution crossover mapping reveals similarities and differences of male and female recombination in maize
High-resolution crossover mapping reveals similarities and differences of male and female recombination in maize
Sex-specific meiotic crossover (CO) landscapes have been identified in multiple species. Here, the authors show that male and female meioses in maize have similar CO landscapes, and differences between COs in the two sexes only exists in their location relative to transcription start sites and some chromatin marks.
- Penny M. A. Kianian
- , Minghui Wang
- & Wojciech P. Pawlowski
-
Article
| Open Access Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia
Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia
There are various indigenous cattle breeds in East Asia which have a complex history. Here, the authors analyse the genomes of 49 modern breeds and eight ancient samples and identify three distinct ancestries and multiple adaptive introgressions from other bovine species.
- Ningbo Chen
- , Yudong Cai
- & Chuzhao Lei
-
Article
| Open Access Gene flow contributes to diversification of the major fungal pathogen Candida albicans
Gene flow contributes to diversification of the major fungal pathogen Candida albicans
The fungal pathogen Candida albicans can undergo a parasexual process that may contribute to genetic diversity, but its actual relevance is unclear. Here, Ropars et al. analyse the genomic sequences of 182 C. albicans isolates collected worldwide and find evidence of gene flow and thus parasexuality in nature.
- Jeanne Ropars
- , Corinne Maufrais
- & Christophe d’Enfert
-
Article
| Open Access High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing
High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing
Functional characterization of disease-causing variants at risk loci in cancer is challenging. Here, in prostate cancer the authors report a pipeline for high-throughput single-nucleotide polymorphisms sequencing (SNPs-seq) for large scale screening of functional SNPs at disease risk loci.
- Peng Zhang
- , Ji-Han Xia
- & Liang Wang
-
Article
| Open Access Population size changes and selection drive patterns of parallel evolution in a host–virus system
Population size changes and selection drive patterns of parallel evolution in a host–virus system
Pathogens exert strong selection on hosts and thus may promote parallel evolution. Here, the authors find that hosts experimentally coevolving with a virus have parallel changes in population size, phenotype, and genomic regions, but accelerated divergence in genome sequence likely due to population size fluctuation.
- Jens Frickel
- , Philine G. D. Feulner
- & Lutz Becks
-
Article
| Open Access Repeated evolution of self-compatibility for reproductive assurance
Repeated evolution of self-compatibility for reproductive assurance
Mating-type switching enables self-compatible reproduction in fungi, but switching ability is variable even within species. Here, the authors find de novo evolution of switching genotypes in experimentally evolved fission yeast populations and show a trade-off between mating success and growth.
- Bart P. S. Nieuwenhuis
- , Sergio Tusso
- & Simone Immler
-
Article
| Open Access Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese
Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese
Recent natural selection left signals in human genomes. Here, Okada et al. generate high-depth whole-genome sequence (WGS) data (25.9×) from 2,234 Japanese people of the BioBank Japan Project (BBJ), and identify signals of recent natural selection which overlap variants associated with human traits.
- Yukinori Okada
- , Yukihide Momozawa
- & Yoichiro Kamatani
-
Article
| Open Access Host-mediated selection impacts the diversity of Plasmodium falciparum antigens within infections
Host-mediated selection impacts the diversity of Plasmodium falciparum antigens within infections
Host immune responses exert selective pressure on Plasmodium falciparum. Here, the authors show that allele-specific immunity impacts the antigenic diversity of individual malaria infections. This process partially explains the extreme amino acid diversity of many parasite antigens and suggests that vaccines should account for allele-specific immunity.
- Angela M. Early
- , Marc Lievens
- & Daniel E. Neafsey
-
Article
| Open Access Population genomics of finless porpoises reveal an incipient cetacean species adapted to freshwater
Population genomics of finless porpoises reveal an incipient cetacean species adapted to freshwater
Whales, dolphins and porpoises are adapted to various aquatic habitats. Here, Zhou et al. show that polymorphisms associated with renal function and the urea cycle have undergone selection in the freshwater Yangtze finless porpoise and provide genomic evidence of incipient speciation.
- Xuming Zhou
- , Xuanmin Guang
- & Guang Yang
-
Article
| Open Access Gene-by-environment interactions in urban populations modulate risk phenotypes
Gene-by-environment interactions in urban populations modulate risk phenotypes
Individuals with different genotypes may respond differently to environmental variation. Here, Favé et al. find substantial impacts of different environment exposures on the transcriptome and clinical endophenotypes when controlling for genetic ancestry by analyzing data from ∼1000 individuals from a founder population in Quebec.
- Marie-Julie Favé
- , Fabien C. Lamaze
- & Philip Awadalla
-
Article
| Open Access Convergent genomic signatures of domestication in sheep and goats
Convergent genomic signatures of domestication in sheep and goats
The sheep and goat were domesticated ~10,500 years ago in the same region of the Middle-East. Here, Alberto et al compare the genomes of wild Asiatic mouflon and Bezoar ibex with that of domestics from local, traditional and improved breeds and find common targets of selection related to domestication and improvement in sheep and goats.
- Florian J. Alberto
- , Frédéric Boyer
- & François Pompanon
-
Article
| Open Access Strong selection during the last millennium for African ancestry in the admixed population of Madagascar
Strong selection during the last millennium for African ancestry in the admixed population of Madagascar
The population of Madagascar arose from admixture of Austronesian and Bantu genetic backgrounds. Analyzing local ancestry in genomes of 700 Malagasy, Pierron et al. identify signals of recent positive selection for African ancestry in a region on chromosome 1 with implications for physiology and disease risk.
- Denis Pierron
- , Margit Heiske
- & Thierry Letellier
-
Article
| Open Access Diverse genetic error modes constrain large-scale bio-based production
Diverse genetic error modes constrain large-scale bio-based production
The declining performance of scale-up bioreactor cultures is commonly attributed to phenotypic and physical heterogeneities. Here, the authors reveal multiple recurring intra-pathway error modes that limit engineered E. coli mevalonic acid production over time- and industrial-scale fermentations.
- Peter Rugbjerg
- , Nils Myling-Petersen
- & Morten O. A. Sommer
-
Article
| Open Access Localization of adaptive variants in human genomes using averaged one-dependence estimation
Localization of adaptive variants in human genomes using averaged one-dependence estimation
Selective sweeps are events in which beneficial mutations spread rapidly through a population. Here, Sugden et al. develop SWIF(r), a probabilistic classification framework for detecting and localizing selective sweeps, and apply it to genomic data from the ‡Khomani San.
- Lauren Alpert Sugden
- , Elizabeth G. Atkinson
- & Sohini Ramachandran
-
Article
| Open Access High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell
High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell
Long-read sequencing technologies facilitate efficient and high quality genome assembly. Here Michael et al. achieve a fast reference assembly for Arabidopsis thaliana KBS-Mac-74 accession using the handheld Oxford Nanopore MinION sequencer and consumer computing hardware, and demonstrate its usefulness in resolving complex structural variation.
- Todd P. Michael
- , Florian Jupe
- & Joseph R. Ecker
-
Article
| Open Access The genetic prehistory of the Baltic Sea region
The genetic prehistory of the Baltic Sea region
The population history of Europe is complex and its very north has not yet been comprehensively studied at a genetic level. Here, Mittnik et al. report genome-wide data from 38 ancient individuals from the Eastern Baltic, Russia and Scandinavia to analyse gene flow throughout the Mesolithic and Bronze Age.
- Alissa Mittnik
- , Chuan-Chao Wang
- & Johannes Krause
-
Article
| Open Access RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis
RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis
Horticultural lettuce varieties vary considerably in phenotype. Here, via RNA-seq of 240 different lettuce accessions, the authors identify loci and expression patterns associated with flavonoid and anthocyanin content and show that cultivated lettuce likely arose via a single domestication event.
- Lei Zhang
- , Wenqing Su
- & Hanhui Kuang
-
Article
| Open Access Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans
Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans
African populations show a high level of genetic diversity and extensive regional admixture. Here, the authors sequence the whole genomes of 24 South African individuals of different ethnolinguistic origin and find substantive genomic divergence between two southeastern Bantu-speaking groups.
- Ananyo Choudhury
- , Michèle Ramsay
- & Michael S. Pepper
-
Article
| Open Access Phenotypic plasticity promotes recombination and gene clustering in periodic environments
Phenotypic plasticity promotes recombination and gene clustering in periodic environments
Selection for recombination requires genetic diversity and negative linkage disequilibrium, which can be produced by coevolutionary arms races. Here the authors propose a qualitatively different scenario that can favour recombination in seasonal environments through the ‘genomic storage effect’.
- Davorka Gulisija
- & Joshua B. Plotkin
-
Article
| Open Access Rapid neo-sex chromosome evolution and incipient speciation in a major forest pest
Rapid neo-sex chromosome evolution and incipient speciation in a major forest pest
The evolution of new sex chromosomes potentially generates reproductive isolation. Here, Bracewell et al. combine crossing experiments with population and functional genomics to characterize neo-sex chromosome evolution and incipient speciation in the mountain pine beetle, Dendroctonus ponderosae.
- Ryan R. Bracewell
- , Barbara J. Bentz
- & Jeffrey M. Good
 Patterns of genetic differentiation and the footprints of historical migrations in the Iberian Peninsula
Patterns of genetic differentiation and the footprints of historical migrations in the Iberian Peninsula