
Featured
-
-
Article |
 Clonally expanded CD8 T cells characterize amyotrophic lateral sclerosis-4
Clonally expanded CD8 T cells characterize amyotrophic lateral sclerosis-4
An immune signature characterized by activated antigen-specific CD8 T cells is identified in the brain and blood of mice with amyotrophic lateral sclerosis-4 (ALS4), suggesting that the immune system is involved in ALS4 neurodegeneration.
- Laura Campisi
- , Shahab Chizari
- & Ivan Marazzi
-
Article
| Open Access Genetic and chemotherapeutic influences on germline hypermutation
Genetic and chemotherapeutic influences on germline hypermutation
A study of 21,879 families with rare genetic diseases identifies 12 with 2- to 7-fold excess of germline mutations, most of which are due to DNA repair defects or exposure to mutagenic chemotherapy, although most individuals with a hypermutated genome will not have a genetic disease.
- Joanna Kaplanis
- , Benjamin Ide
- & Matthew Hurles
-
Article |
 Evidence for 28 genetic disorders discovered by combining healthcare and research data
Evidence for 28 genetic disorders discovered by combining healthcare and research data
By integrating healthcare and exome-sequencing data from parent–offspring trios of patients with developmental disorders, 28 genes that had not previously been associated with developmental disorders were identified.
- Joanna Kaplanis
- , Kaitlin E. Samocha
- & Kyle Retterer
-
Article |
 Genome-wide detection of tandem DNA repeats that are expanded in autism
Genome-wide detection of tandem DNA repeats that are expanded in autism
Genome-wide analysis of tandem DNA repeats in the genomes of individuals with autism spectrum disorder and control participants reveals a strong contribution of tandem repeat expansions to the genetic aetiology and phenotypic complexity of autism spectrum disorder.
- Brett Trost
- , Worrawat Engchuan
- & Ryan K. C. Yuen
-
Article |
 MeCP2 links heterochromatin condensates and neurodevelopmental disease
MeCP2 links heterochromatin condensates and neurodevelopmental disease
The chromatin protein MeCP2 is a component of dynamic, liquid-like heterochromatin condensates, and the ability of MeCP2 to form condensates is disrupted by mutations in the MECP2 gene that occur in the neurodevelopmental disorder Rett syndrome.
- Charles H. Li
- , Eliot L. Coffey
- & Richard A. Young
-
Letter |
 Common genetic variants contribute to risk of rare severe neurodevelopmental disorders
Common genetic variants contribute to risk of rare severe neurodevelopmental disorders
A genome-wide association study of approximately 7,000 patients with neurodevelopmental disorders demonstrates that overall risk and clinical presentation in putative monogenic disorders is also influenced by common genetic variants present in the general population.
- Mari E. K. Niemi
- , Hilary C. Martin
- & Jeffrey C. Barrett
-
Article |
 De novo mutations in regulatory elements in neurodevelopmental disorders
De novo mutations in regulatory elements in neurodevelopmental disorders
Analysis of rare de novo mutations in gene regulatory elements suggests that 1–3% of patients with neurodevelopmental disorders carry such mutations in elements that are active in the fetal brain.
- Patrick J. Short
- , Jeremy F. McRae
- & Matthew E. Hurles
-
Letter |
 Kctd13 deletion reduces synaptic transmission via increased RhoA
Kctd13 deletion reduces synaptic transmission via increased RhoA
Experimental evidence that global Kctd13 reduction leads to increased RhoA levels that reduce synaptic transmission, implicating RhoA as a potential therapeutic target for neuropsychiatric disorders associated with copy-number variants that include KCTD13.
- Christine Ochoa Escamilla
- , Irina Filonova
- & Craig M. Powell
-
Letter |
 Radically truncated MeCP2 rescues Rett syndrome-like neurological defects
Radically truncated MeCP2 rescues Rett syndrome-like neurological defects
Analysis of the minimal functional unit for MeCP2 protein shows that its function is to recruit the NCoR/SMRT co-repressor complex to methylated sites on chromatin, which may have use in designing strategies for gene therapy of Rett syndrome.
- Rebekah Tillotson
- , Jim Selfridge
- & Adrian Bird
-
Letter |
 A human neurodevelopmental model for Williams syndrome
A human neurodevelopmental model for Williams syndrome
A human neurodevelopmental model fills the current knowledge gap in the cellular biology of Williams syndrome and could lead to further insights into the molecular mechanism underlying the disorder and the human social brain.
- Thanathom Chailangkarn
- , Cleber A. Trujillo
- & Alysson R. Muotri
-
Letter |
 Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice
Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice
Deep brain stimulation (DBS) of the fimbria–fornix—a region that provides input to the hippocampus—is shown to restore hippocampus-dependent memory and hippocampal long-term potentiation and neurogenesis in a mouse model of Rett syndrome, suggesting that DBS, which is already used in the treatment of several neurological conditions, could be a viable approach to mitigating cognitive impairment in Rett syndrome and other disorders of childhood intellectual disability.
- Shuang Hao
- , Bin Tang
- & Jianrong Tang
-
Brief Communications Arising |
 Wild-type microglia do not reverse pathology in mouse models of Rett syndrome
Wild-type microglia do not reverse pathology in mouse models of Rett syndrome
- Jieqi Wang
- , Jan Eike Wegener
- & Andrew A. Pieper
-
Letter |
 Large-scale discovery of novel genetic causes of developmental disorders
Large-scale discovery of novel genetic causes of developmental disorders
Up to half of children with severe developmental disorders of probable genetic origin remain without a genetic diagnosis; here, in a systematic and nationwide study of 1,133 children with severe, undiagnosed developmental disorders, and their parents, exome sequencing and array-based detection of chromosomal rearrangements reveals novel genes causing developmental disorders, increasing the proportion of children that can now be diagnosed to 31%.
- T. W. Fitzgerald
- , S. S. Gerety
- & M. E. Hurles
-
Letter |
 Towards a therapy for Angelman syndrome by targeting a long non-coding RNA
Towards a therapy for Angelman syndrome by targeting a long non-coding RNA
Angelman syndrome is a neurodevelopmental disorder caused by disrupted function of the maternal copy of the imprinted UBE3A gene; here, targeting a long non-coding RNA that is responsible for silencing the paternal copy of UBE3A with antisense oligonucleotides is shown to partially restore UBE3A expression in the central nervous system and correct some cognitive deficits in a mouse model of the disease.
- Linyan Meng
- , Amanda J. Ward
- & Frank Rigo
-
Article |
 The contribution of de novo coding mutations to autism spectrum disorder
The contribution of de novo coding mutations to autism spectrum disorder
Family-based exome sequencing in a large autism study has identified 27 high-confidence gene targets and accurately estimates the contribution of both de novo gene-disrupting and missense mutations to the incidence of simplex autism, with target genes in affected females overlapping those in males of lower but not higher IQ; targets also overlap known targets for intellectual disability and schizophrenia, and are enriched for chromatin modifiers, FMRP-associated genes and embryonically expressed genes.
- Ivan Iossifov
- , Brian J. O’Roak
- & Michael Wigler
-
-
-
Letter |
 Genome sequencing identifies major causes of severe intellectual disability
Genome sequencing identifies major causes of severe intellectual disability
Whole-genome sequencing is used to identify genetic alterations in patients with severe intellectual disability for whom all other tests, including array and exome sequencing, returned negative results; de novo single-nucleotide and copy number variations affecting the coding region seem to be a major cause of this disorder.
- Christian Gilissen
- , Jayne Y. Hehir-Kwa
- & Joris A. Veltman
-
Letter |
 De novo mutations in epileptic encephalopathies
De novo mutations in epileptic encephalopathies
Exome sequencing has found an excess of de novo mutations in the ∼4,000 most intolerant genes in patients with two classical epileptic encephalopathies (infantile spasms and Lennox–Gastaut syndrome); among them are multiple de novo mutations in GABRB3 and ALG13.
- Andrew S. Allen
- , Samuel F. Berkovic
- & Melodie R. Winawer
-
News & Views |
Signalling pathways of fragile X syndrome
The RNA-binding protein FMR1 has a key role in the neurodevelopmental disorder fragile X syndrome, but the RNAs targeted by the protein were mostly unknown. An analysis reveals thousands of possible targets. See Article p.382
- Sabarinath Jayaseelan
- & Scott A. Tenenbaum
-
Letter |
 KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant
KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant
Overexpression of all 29 human transcripts of a region of the 16p11.2 chromosome in zebrafish embryos identifies KCTD13 as the message inducing the microcephaly phenotype associated with 16p11.2 duplication, whereas its suppression yields the macrocephalic phenotype associated with the reciprocal deletion, suggesting that KCTD13 is a major driver for the neurodevelopmental phenotypes associated with the 16p11.2 copy number variants.
- Christelle Golzio
- , Jason Willer
- & Nicholas Katsanis
-
News |
 Gene hunt is on for mental disability
Gene hunt is on for mental disability
Pioneering clinical genome-sequencing projects focus on patients with developmental delay.
- Ewen Callaway
-
Research Highlights |
Mutation and infection to blame
-
News & Views |
 Drugs to awaken a paternal gene
Drugs to awaken a paternal gene
Mutations in the maternal copy of the UBE3A gene cause a neurodevelopmental disorder known as Angelman syndrome. Drugs that activate the normally silenced paternal copy of this gene may be of therapeutic value. See Letter p.185
- Arthur L. Beaudet
-
News |
 A wake-up call for dormant genes
A wake-up call for dormant genes
Anti-cancer drug holds potential as a treatment for genetic-imprinting disorder.
- Rebecca Hill
-
Letter |
 Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons
Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons
Cancer drugs that can potentially treat Angelman syndrome are identified.
- Hsien-Sung Huang
- , John A. Allen
- & Benjamin D. Philpot
-
News |
 Pollutants' role in birth defects becomes clearer
Pollutants' role in birth defects becomes clearer
Levels of polycyclic aromatic hydrocarbons linked to neural tube defects.
- Katharine Sanderson
-
Research Highlights |
Defects from stunted neurites
-
News |
 Disease-in-a-dish approach gives clues to Rett syndrome
Disease-in-a-dish approach gives clues to Rett syndrome
Unregulated jumping genes are a possible culprit for the debilitating disease.
- David Cyranoski
-
Letter |
 L1 retrotransposition in neurons is modulated by MeCP2
L1 retrotransposition in neurons is modulated by MeCP2
Long interspersed nuclear elements-1 (L1) retrotransposons affect gene expression and neuronal function throughout brain development. These authors show that the absence of methyl-CpG-binding protein 2, a modulator of DNA methylation implicated in several neurodevelopmental disorders, increases L1 retrotransposon activity in rodent models, with this increase in susceptibility duplicated in patients with Rett syndrome. These correlations suggest that disease-related genetic mutations may influence L1 retrotransposon activity.
- Alysson R. Muotri
- , Maria C. N. Marchetto
- & Fred H. Gage
-
Article |
 Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes
Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes
Mutations in the methyl-CpG-binding protein 2 (MeCP2) gene cause Rett syndrome, a neurodevelopmental disorder with features of autism. Multiple mouse models of MeCP2 have been generated, but show only a subset of the symptoms of Rett syndrome. These authors find that mice with selective deletion of MeCP2 in GABA-mediated neurons show not only impaired GABA-mediated function, but capitulate multiple key features of Rett, further suggesting a role of inhibitory function in neuropsychiatric disease.
- Hsiao-Tuan Chao
- , Hongmei Chen
- & Huda Y. Zoghbi
-
Letter |
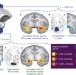 Amygdalar and hippocampal substrates of anxious temperament differ in their heritability
Amygdalar and hippocampal substrates of anxious temperament differ in their heritability
Anxious temperament in both humans and monkeys is an important early predictor of psychopathology and is known to be heritable. These authors characterize the neural circuitry associated with this trait and the extent to which its function is heritable. A scan of related monkeys after exposure to mild stress showed that activation in both the amygdala and hippocampus was predictive of anxious temperament, but that heritability of activity in hippocampus was greater than that in amygdala.
- Jonathan A. Oler
- , Andrew S. Fox
- & Ned H. Kalin
-
News |
 Children who form no racial stereotypes found
Children who form no racial stereotypes found
Brain disorder eradicates ethnic but not gender bias.
- Janelle Weaver
 Polygenic architecture of rare coding variation across 394,783 exomes
Polygenic architecture of rare coding variation across 394,783 exomes Bias towards large genes in autism
Bias towards large genes in autism