


















« Prev Next »
The cloning of expressed genes and the polymerase chain reaction (PCR), two biotechnological breakthroughs of the 1970s and 1980s, continue to play significant roles in science today. Both technologies give researchers the means to make more DNA, but they do so in different ways. In particular, cloning involves the synthesis of DNA from mRNA using an enzyme called reverse transcriptase. Although this method reverses the flow of genetic information as described by the central dogma, it effectively mimics the process by which RNA viruses "flip" the direction of transcription in their host cells, thereby causing these cells to manufacture viral DNA even though the viruses themselves contain only RNA. In contrast, the polymerase chain reaction does not involve the use of an initial mRNA template to manufacture DNA. Rather, PCR involves the synthesis of multiple copies of specific DNA fragments using an enzyme known as DNA polymerase. This method allows for the creation of literally billions of DNA molecules within a matter of hours, making it much more efficient than the cloning of expressed genes. However, cloning remains the go-to method for researchers when only the mRNA template (and not the DNA template) of a sequence of interest is available.
Making DNA from RNA: Reversal of the Central Dogma
"The central dogma, enunciated by Crick in 1958 and the keystone of molecular biology ever since, is likely to prove a considerable over-simplification. That is the heretical but inescapable conclusion stemming from experiments done in the past few months in two laboratories in the United States."
--"Central Dogma Reversed," Nature, June 27, 1970
The so-called central dogma of molecular biology states that all genetic information flows in one direction: from DNA to RNA through the process of transcription, and then from RNA to protein through the process of translation (Crick, 1958). For over a decade, the central dogma was thought to be a universal truth--in other words, researchers believed that genetic information always flowed in this order, otherwise it could not be passed along. In 1970, however, the two experiments mentioned in the Nature quote--one conducted by David Baltimore, then of the California Institute of Technology in Pasadena, and the other by Howard Temin and Satoshi Mizutani, then of the University of Wisconsin in Madison--called this belief into question. Specifically, these researchers independently published scientific papers demonstrating that RNA tumor viruses contain enzymes that use viral RNA as a template for the synthesis of DNA, thereby reversing the direction of transcription (Baltimore, 1970; Temin & Mizutani, 1970). Not only did these two experiments challenge the validity of the central dogma, but they also laid the foundation for a series of technological developments that eventually earned reverse transcription and the synthesis of complementary DNA, or cDNA, central places in the molecular biologist's toolbox.
Discovering Reverse Transcription
During the late 1960s, Baltimore, Temin, and Mizutani were each driven by unanswered questions about how RNA viruses transformed healthy cells into tumor cells. They knew that transformation ensued when healthy cells incorporated DNA from the external environment (in this case, RNA tumor virus DNA) into their genomes. But how could a eukaryotic cell incorporate DNA from a virus that didn't have any DNA?
Howard Temin had hypothesized the existence of an enzyme capable of making DNA from RNA as early as 1964 ("Central Dogma Reversed," 1970). But, as is the case with all scientific hypotheses, the research community remained skeptical of this proposal until the 1970 publications wiped that skepticism away. At that point, the race was on to identify the enzyme responsible for the creation of DNA from RNA. Today, that enzyme is known as reverse transcriptase.
Interestingly, in their groundbreaking papers, the two sets of scientists didn't actually identify reverse transcriptase, but they did provide clear and conclusive evidence of the existence of an enzyme that utilized viral RNA as a template for DNA synthesis. The experiments supporting the existence of this DNA polymerase produced data that revealed the following:
- The DNA polymerase only incorporated deoxyribonucleotides, not ribonucleotides, into its product.
- The product itself "behaved" like DNA--in other words, it was sensitive to treatment by deoxyribonucleases but not ribonucleases.
- The RNA itself was the template, as shown by the fact that treatment of virions with ribonucleases destroyed the ability of the polymerase to incorporate radioactively labeled nucleotides.
Although the motivation for both studies was to better understand the role of viruses in some cancers, there is also some suggestion in the papers that the scientists were aware, at least on an intuitive level, that there were far greater implications to their findings. As Temin and Mizutani (1970) wrote, "This result would have strong implications for theories of viral carcinogenesis and, possibly, for theories of information transfer in other biological systems."
It did not take long for scientists to isolate the reverse transcriptase responsible for Baltimore's findings (Verma et al., 1972). Another team (Bank et al., 1972) then used the enzyme to synthesize DNA from mRNA in a test tube for the first time. (The so-called complementary DNA that results is referred to as cDNA.) Both teams used globin mRNAs, or mRNAs that encode blood hemoglobin polypeptides, to demonstrate that reverse transcriptase does in fact synthesize DNA from mRNA templates. Moreover, the teams also found that the reaction works best in the abundance of short sequences composed entirely of thymine nucleotides known as oligo(dT) primers. Knowing that most eukaryotic mRNAs have a string of adenine nucleotides--also known as a poly(A) tail--at their 3′ end, the scientists had predicted that cDNA synthesis would require oligo(dT) primers, or that it would at least be made more efficient by the presence of these primers.
How Reverse Transcriptase Works
So how exactly does reverse transcriptase work? As shown in Figure 1, the production of cDNA involves two basic steps: separating mRNA molecules from other cellular RNA, and then using reverse transcriptase to copy the mRNA molecules into cDNA.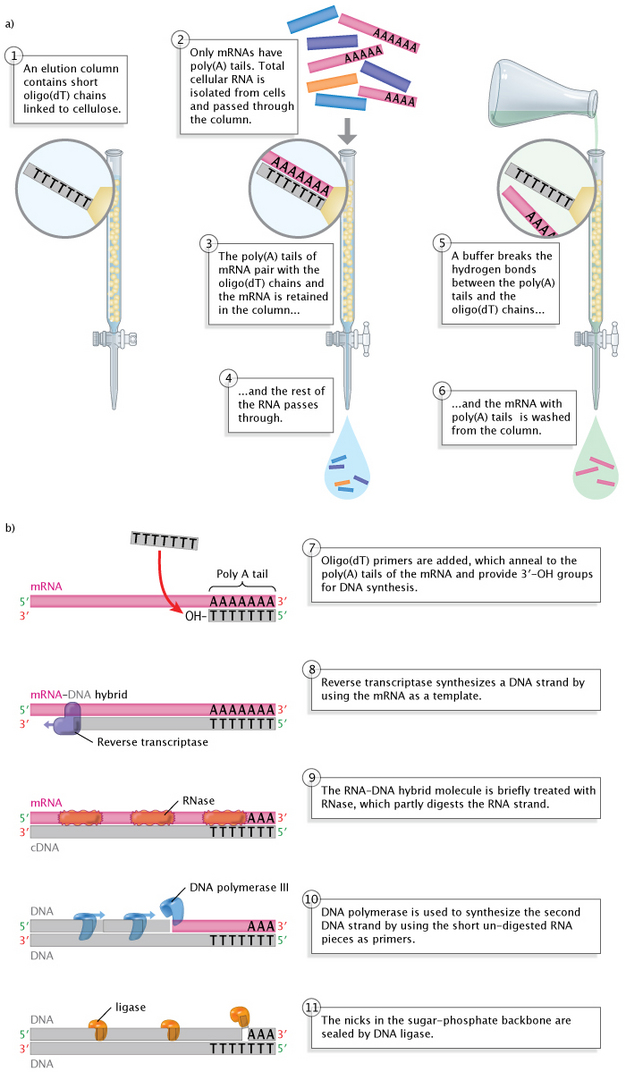
Figure 1: The production of cDNA from mRNA.
cDNA is produced in two basic steps: (a) first, mRNA is isolated from other cellular RNA using an elution column. (b) Second, the enzyme reverse transcriptase synthesizes strands of DNA using the mRNA molecules as templates.
© 2014 Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved. 
The first step relies on the fact that most eukaryotic mRNAs have poly(A) tails at their 3′ ends. The poly(A) tails serve as "hooks" during the separation process. This process involves pouring all of the cellular RNA (tRNA, rRNA, snRNA, mRNA, etc.) into what is known as an elution column of short DNA pieces, mostly thymine nucleotides. As the RNA mixture moves downward through the column, the poly(A) tails of the mRNA molecules bind to the thymine nucleotides. The rest of the RNA molecules--those without poly(A) tails, in other words--run right through. Afterward, the column is washed with a solution that breaks the hydrogen A-T bonds, and the released mRNA molecules are collected.
The second step, the copying of the now-isolated mRNA molecules into cDNA, involves adding oligo(dT) primers to the mRNA collection. These primers pair with the poly(A) tails of the mRNA molecules, again at the 3′ end, providing the exposed 3′-OH group required for the initiation of DNA synthesis. At this point, the reverse transcriptase enzyme is added, and this enzyme proceeds to utilize the mRNA strand as a template for the synthesis of a complementary DNA strand. The enzyme adds nucleotides, one by one, to the 3′ end of the new strand, with each newly added nucleotide a complement to its template pair just as in DNA replication, with the exception that RNA contains Us in place of Ts. Thus, Cs are paired with Gs and vice versa, Ts are paired with As, and As are paired with Us. Scientists then use several different methods to convert the RNA-DNA hybrids into double-stranded cDNA molecules, such as enzymatic digestion of the RNA strand followed by DNA synthesis utilizing, this time, the cDNA strand as the template.
Using Reverse Transcriptase to Clone Expressed Genes
Following the development of this method, the use of reverse transcriptase to clone expressed genes grew for several decades. However, there were limits to this practice. For example, most cDNA molecules that were synthesized in a single reaction were incomplete, with the 5′ end of the mRNA not represented in the final cDNA. Therefore, the cDNA did not contain a complete copy of the amino acid-encoding region of the gene.
Eventually, in the late 1990s, Piero Carninci and his colleagues at the Genome Science Laboratory in Ibaraki, Japan, devised a series of methods to get around this and other problems. In particular, these researchers developed a new technique for selecting full-length cDNA molecules. This process is known as biotin capping, and it involves capping the 5′ end of the mRNA with a biotin group and then washing the cDNAs with an RNA digestion enzyme, like RNAse I. When washed with a solution containing a cap-binding protein, all of the cDNA-mRNA hybrids with only partial cDNA copies are carried away, thus only leaving behind the hybrids with full-length cDNA molecules. In fact, the researchers demonstrated that biotin capping yielded about 95% full-length cDNA clones (Carninci et al., 1996).
Today, scientists continue to build and utilize what are known as cDNA libraries, or collections of cDNAs from particular tissues gathered at particular times during an organism's life cycle. The synthesis of cDNA molecules is referred to as cloning, because the cDNA molecules are matching copies of the DNA responsible for encoding the mRNA template.
Scientists often generate cDNA libraries as a way to find genes of interest. They screen these libraries using what are known as probes--complementary pieces of DNA that hybridize to the cDNA molecules. They also use cDNA libraries to identify genes that are expressed differently in different types of tissues or at different developmental stages. Libraries of cDNA molecules provide snapshots of gene activity, because only those genes that are actually expressed and transcribed into mRNA molecules can be cloned. For example, one would expect a cDNA library compiled from mRNA isolated during a stage of prenatal development to be very different from a cDNA library generated from sequences transcribed during adulthood.
Making Copies via Polymerase Chain Reaction
Arguably, the polymerase chain reaction (PCR) machine has recently become as indispensible to biological research as the light microscope was some 100 years ago. Like gene expression and cloning, the idea of PCR was born only in the early 1970s (Swaminathan, 2007). It would take more than a decade before American biochemist Kary Mullis turned the idea into reality (Mullis & Faloona, 1987). Until Mullis's success with this method, the only way biologists could make copies of whatever gene they were interested in was by the relatively laborious and time-consuming process of identifying and isolating the gene--in other words, through constructing and screening a cDNA library, as described earlier--and then inserting that gene into living cells that replicated the target DNA along with their own DNA. In contrast, PCR enables the production, or amplification, of billions of copies of an original piece of DNA in a test tube within minutes or hours, not days.
How PCR Works
Basically, PCR is DNA replication on a grander scale. The polymerase chain reaction relies on the use of several essential chemical ingredients, including the following:
- A DNA polymerase
- A small amount of DNA to serve as the initial template
- The four deoxyribonucleotides to serve as the substrates for the DNA polymerase and the raw ingredients of the new DNA molecules
- A few necessary ions and salts
- A pair of primers with exposed 3′-OH groups that will bind to the particular sequence of interest in the DNA template
As previously mentioned, the DNA polymerases can only add new nucleotides to the 3′-OH end of a growing strand. They therefore require the presence of a primer to get started, because they cannot begin synthesis de novo. In fact, two primers are required--one to initiate replication of each of the two DNA strands.
A single PCR reaction involves three temperature-dependent steps, described as follows:
- The starting solution is heated, usually to between 90° and 100°C. The high temperatures break the hydrogen bonds between the two strands of the original DNA double helix, providing the necessary single-stranded templates.
- After just a couple of minutes at that temperature, the reaction mixture is quickly cooled, usually to somewhere between 30° and 65°C. It is then held for less than a minute at this lower temperature--which is enough time for the primers to bind to their complementary sequences on the single-stranded templates.
- The sample is next heated to 60° to 75°C for less than a minute, during which time the DNA polymerase adds nucleotides to the primer, synthesizing a new DNA strand using only the template sequences that bind the primers (Figure 2).

Figure 2: The polymerase chain reaction (PCR).
Two key innovations facilitated the use of PCR in the laboratory: the discovery of a DNA polymerase that is stable at the high temperatures used in step 1 of PCR and the development of automated thermal cyclers (machines that bring about the rapid temperature changes necessary for the different steps of PCR).
© 2014 Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved.
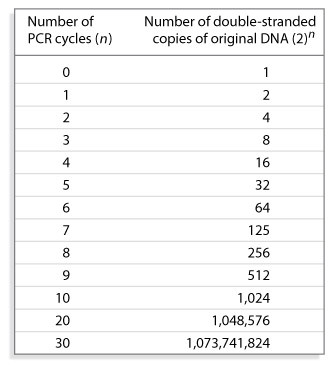
Figure 3: The number of double-stranded copies of original DNA produced over the course of 30 PCR cycles.
One molecule of DNA increases to more than a thousand molecules in ten PCR cycles, to more than one million molecules in twenty cycles, and to more than one billion molecules in thirty cycles.
© 2014 Nature Education Adapted from Pierce, Benjamin. Genetics: A Conceptual Approach, 2nd ed. All rights reserved.
The many changes in temperature required during multiple PCR cycles are carried out in a thermocycler, also known as a PCR machine. After PCR cycling is complete, the amplification products can be subjected to cloning, sequencing, or analysis via gel electrophoresis.
Advances in PCR Technology
As with all genetic technologies, of course, scientists have improved and refined the original PCR process described by Mullis and Faloona in 1987. For example, one of the major limitations of early PCR methods was that fresh DNA polymerase had to be added during every cycle. This repetitive step was not just tedious, but it also greatly increased the likelihood of error. Mullis and colleagues addressed this deficiency just a year later when they demonstrated how a particular type of DNA polymerase, a heat-resistant enzyme isolated from the bacterium Thermus aquaticus, eliminated the need to add fresh polymerase during every cycle. Thermus aquaticus--often referred to by its popular nickname "Taq polymerase"--is a thermophilic bacterium that can survive temperatures up to 95°C. In fact, its natural habitat is the hot spring ecosystem of Yellowstone National Park. This innovation greatly improved the quantity and quality of PCR products (Saiki et al., 1988).
More recently, another major PCR innovation was the development of real-time PCR. This refinement involves the use of dyes or fluorescent probes that eliminate the need for post-PCR electrophoresis. In real-time PCR, the fluorescence that is associated with the accumulation of newly amplified DNA is measured through the use of an optical sensing system.
Summary
Thus, both cloning of expressed genes and PCR continue to serve as essential tools for genetic researchers. Cloning--which involves the creation of DNA from mRNA and thus represents a reversal of the central dogma--is particularly useful when scientists aren't able to isolate the DNA template of a sequence of interest. In addition, because this method relies on mRNA rather than DNA, it provides an excellent means for studying the differences in gene expression in different cells at different points in development. PCR, on the other hand, is more akin to "traditional" DNA synthesis in that it requires the presence of an initial DNA (rather than RNA) template. The primary advantage of PCR is its speed--even if researchers begin with only a single segment of DNA, they can produce literally billions of molecules within a matter of hours. As with all technologies, scientists continue to improve both PCR and the cloning process, thereby ensuring that these methods will play a role in genetic breakthroughs for years to come.
References and Recommended Reading
Baltimore, D. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226, 1209–1211 (1970) doi:10.1038/2261209a0 (link to article)
Bank, A., et al. In vitro synthesis of DNA components of human genes for globins. Nature New Biology 235, 167–169 (1972)
Carninci, P., et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 37, 327–336 (1996)
Central dogma reversed. Nature 226, 1198–1199 (1970) doi:10.1038/2261198a0 (link to article)
Crick, F. On protein synthesis. The Biological Replication of Macromolecules: Symposium for the Society of Experimental Biology 12, 138–162 (1958)
Mullis, K. B., & Faloona, F. A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods in Enzymology 155, 335–350 (1987)
Saiki, R. K., et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487–491 (1988) doi:10.1126/science.2448875
Swaminathan, S. Milestone 11: Chain reaction. Nature.com, (2007) (accessed July 3, 2008)
Temin, H. M., & Mizutani, S. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of rous sarcoma virus. Nature 226, 1211–1213 (1970) doi:10.1038/2261211a0 (link to article)
Verma, I. M., et al. In vitro synthesis of DNA complementary to rabbit reticulocyte 10s RNA. Nature New Biology 235, 163–167 (1972).










