


 Figure 1: Structural variation map.
Figure 1: Structural variation map.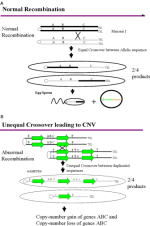
















« Prev Next »
Analysis of individual human genomes has revealed an unexpected amount of variability in human populations. The most common form of genetic variation involves small changes in the genetic code that alter a single base pair. Other types of mutations range from small insertions to large chromosomal rearrangements that can be detected cytogenetically using a microscope. The term "copy number variation" refers to an intermediate-scale genetic change, operationally defined as segments greater than 1,000 base pairs in length but typically less than 5 megabases, which is the cytogenetic level of resolution. CNVs include both additional copies of sequence (duplications) and losses of genetic material (deletions). Because CNVs change the structure of the genome, such mutations, together with inversions and translocations, are collectively classified as forms of genome structural variation. Recently, scientists have come to appreciate that CNVs account for much of human variability.
Copy Number Variation Is Common in Human Genomes

© 2005 Nature Publishing Group Tuzun, E. et al. Fine-scale structural variation of the human genome. Nature Genetics 37, 728. All rights reserved. 
In 2004, researchers published two landmark studies of healthy individuals that showed that copy number changes involving hundreds of thousands of DNA base pairs were unexpectedly common throughout the human genome (Iafrate et al., 2004, Sebat et al., 2004). Subsequent studies using more sensitive methods extended these results, providing evidence for subtle variation over large portions of the human genome. For instance, Figure 1 summarizes the results of one study that compared the sequence of euchromatic regions from one human female with a reference human genome sequence (Tuzun et al., 2005). (The reference sequence is thought to largely represent the sequence of one human male.) This particular study identified 297 sites of potential structural variation between the two sequences, including 139 insertions, 102 deletions, and 56 inversions. The positions of these structural variations are shown for each chromosome.
Based on these and other results (Redon et al., 2006, Kidd et al., 2008; Conrad et al. 2006, McCarroll et al., 2006), human CNV is now thought to affect more base pairs than other forms of mutation. In other words, if two humans were compared, the number of base pairs affected by structural differences in the organization and copy number status of DNA segments would be greater than the sum of all single-base-pair substitution differences. Recent evidence also suggests that changes in copy number play an important role in evolution. For example, a comparison of the human and chimpanzee genetic sequences found that copy number changes account for more differences between the two species than do other forms of mutation (Cheng et al., 2005). Research also reveals that copy number changes can affect the expression of genes, alter the organization of chromatin, and/or influence the regulation of genes in the vicinity.
CNVs Are Generally Placed into Two Categories
Scientists generally assign CNVs to one of two main categories, based on the length of the affected sequence. The first category includes copy number polymorphisms (CNPs), which are common in the general population, occurring with an overall frequency of greater than 1%. CNPs are typically small (most are less than 10 kilobases in length), and they are often enriched for genes that encode proteins important in drug detoxification and immunity. A subset of these CNPs is highly variable with respect to copy number. As a result, different human chromosomes can have a wide range of copy numbers (e.g., 2, 3, 4, 5, etc.) for a particular set of genes. CNPs associated with immune response genes have recently been associated with susceptibility to complex genetic diseases, including psoriasis (Hollox et al., 2008), Crohn's disease (Fellermann et al., 2006), and glomerulonephritis (Aitman et al., 2006).
The second class of CNVs includes relatively rare variants that are much longer than CNPs, ranging in size from hundreds of thousands of base pairs to over 1 million base pairs in length. Also known as microdeletions and microduplications, these variants usually have a much more recent origin within a family. These CNVs may have arisen during production of the sperm or egg that gave rise to a particular individual, or they may have been passed down for only a few generations within a family. These large and rare structural variants have been observed disproportionately in patients with mental retardation, developmental delay, schizophrenia, and autism (de Vries et al., 2005, Sharp et al. 2006, Sebat et al., 2007, Walsh et al., 2008). Their appearance in such patients has led to speculation that large and rare CNVs may be more important in neurocognitive diseases than other forms of inherited mutations, including single nucleotide substitutions.
The Mechanistic Basis for Copy Number Variation
Although the mechanism underlying copy number variation is not completely understood, the fact that both forms of CNV preferentially occur near or within duplicated sequences (Figure 1) has provided some important clues to their origins. During meiosis, maternal and paternal chromosomes normally align along the metaphase plate using sequence homology as a guide to pair and initiate recombination (Figure 2A). The presence of duplicated sequences, however, can "trick" the recombination machinery to initiate a crossover event (Figure 2B) where it normally would not occur. As a result of this aberrant recombination event, known as nonallelic homologous recombination, copies of the duplicated sequence are gained or lost (Lupski, 1998). If the two duplicated copies are separated by unique sequence, this intervening unique sequence can also become a CNV. As compared to other sequenced mammalian genomes, the human genome has a disproportionately larger fraction of these duplicated regions, known as interspersed segmental duplications, which suggests that the human genome is particularly prone to rare and recurrent copy number variation. (It is important to note, however, that not all CNVs arise through this mechanism. Alternative mechanisms are needed to explain the origin of those CNVs whose breakpoints do not map to segmental duplications.)
Implications of Copy Number Variation for Heredity and Disease
Copy number variations have a number of important implications. First, CNVs suggest that an individual's genetic code may not simply be the sum of the genetic contributions of the individual's two parents. Because the unequal crossover events responsible for CNVs occur during the production of sperm and eggs, children may have lost or gained additional copies of genetic information that were present in either of their parents' chromosomes. Second, the extent of CNV and its association with disease has led human geneticists to consider an alternate paradigm for the genetic basis of human diseases. Instead of considering disease to be largely the result of common genetic variants that have been inherited for countless generations, geneticists now recognize that large, rare structural variants of recent origin may provide the genetic basis for common diseases such as mental retardation, autism, and schizophrenia. Although these structural variants may be individually rare, collectively they may be quite common, accounting for a greater proportion of the heritable risk of disease than previously assumed. In total, these data imply that the structure of the human genome is much more dynamic and malleable than previously anticipated.
References and Recommended Reading
Aitman, T. J. et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 439, 851-5 (2006) (link to article)
Bailey, J. A., et al. Recent segmental duplications in the human genome. Science 297, 1003-1007 (2002)
Cheng, Z., et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature 437, 88-93 (2005) doi:10.1038/nature04000 (link to article)
Conrad, D. F., Andrews, T. D., Carter, N. P., Hurles, M. E. & Pritchard, J. K. A high-resolution survey of deletion polymorphisms in the human genome. Nat Genet 38, 75-81 (2006) (link to article)
de Vries, B. B. et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet 77, 606-16 (2005)
Fellermann, K. et al. A chromosome 8 gene-cluster polymorphism with low human Beta-defensin 2 gene copy number predisposes to crohn disease of the colon. Am J Hum Genet 79, 439-48 (2006)
Hollox, E. J. et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet 40, 23-5 (2008) (link to article)
Iafrate, A. J., et al. Detection of large-scale variation in the human genome. Nature Genetics 36, 949-951 (2004) doi:10.1038/ng1416 (link to article)
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860-921 (2001) (link to article)
Kidd, J. M., et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56-64 (2008) doi:10.1038/nature06862
Lupski, J. R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends in Genetics 14, 417-422 (1998)
McCarroll, S. A. et al. Common deletion polymorphisms in the human genome. Nat Genet 38, 86-92 (2006).
Redon, R., et al. Global variation in copy number in the human genome. Nature 444, 444-454 (2006) doi:10.1038/nature05329 (link to article)
Sebat, J., et al. Large-scale copy number polymorphism in the human genome. Science 305, 525-528 (2004)
Sebat, J., et al. Strong association of de novo copy number mutations with autism. Science 316, 445-449 (2007)
Sharp, A. J., et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nature Genetics 38, 1038-1042 (2006) doi:10.1038/ng1862 (link to article)
Stranger, B. E., et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848-853 (2007)
Tuzun, E., et al. Fine-scale structural variation of the human genome. Nature Genetics 37, 727-732 (2005) doi:10.1038/ng1562 (link to article)
Walsh, T., et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539-543 (2008)









