


 Figure 2
Figure 2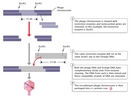
















« Prev Next »

Figure 1: The multicolored GloFish®.
Courtesy of www.glofish.com. All rights reserved. 
"Seeing is believing with GloFish. They are absolutely stunning!" The preceding is some of the marketing material you'd read if you visited the GloFish website (GloFish, 2008). Beauty may be in the eye of the beholder, but nearly everyone would agree that these first—and, so far, only—transgenic animals made available to the general public in the United States (except in California, pending a formal review of their potential effect on the environment) are a worthy conversation piece. A transgenic, or genetically modified, organism is one that has been altered through recombinant DNA technology, which involves either the combining of DNA from different genomes or the insertion of foreign DNA into a genome. GloFish (Figure 1) are a type of transgenic zebrafish (Danio rerio) that have been modified through the insertion of a green fluorescent protein (gfp) gene. Not all GloFish are green, however. Rather, there are several gfp gene constructs, each encoding a different colored phenotype, from fluorescent yellow to fluorescent red.
Currently, GloFish are the only recombinant-DNA animal that has been approved for human "use" by the U.S. Food and Drug Administration. Their approval has raised important questions about whether, and to what extent, genetically modified animals should be made available to consumers. But how were scientists able to create these engineered organisms in the first place? Like so many genetic technologies used today, recombinant DNA technology had its origins in the late 1960s and early 1970s. By the 1960s, scientists had already learned that cells repair DNA breaks by reuniting, or recombining, the broken pieces. Thus, it was only a matter of time before researchers identified the raw biological ingredients necessary for recombination, figured out how those ingredients functioned together, and then tried to govern the recombining process themselves.
Early Experiments Provide the Basis for Recombinant Organisms
Although recombinant DNA technology first emerged in the 1960s and 1970s, the basic principle of recombination had been discovered many years earlier. Indeed, in 1928, Frederick Griffith, an English medical officer studying the bacteria responsible for a pneumonia epidemic in London, first demonstrated what he termed "genetic transformation"; here, living cells took up genetic material released by other cells and became phenotypically "transformed" by the new genetic information. More than a decade later, Oswald Avery repeated Griffith's work and isolated the transforming molecule, which turned out to be DNA. These experiments showed that DNA can be transferred from one cell to another in the laboratory, thus changing the actual genetic phenotype of an organism.
Prior to these classic experiments, the idea that the genetic material was a specific chemical that could be modified and transferred into cells was certainly controversial. But before the explosion in recombinant DNA could begin, scientists would have to learn not only how to transfer DNA, but also how to isolate and modify individual genes.
Key Developments in Recombinant DNA Technology
Following these early experiments, four key developments helped lead to construction of the first recombinant DNA organism (Kiermer, 2007). The first two developments revolved around how scientists learned to cut and paste pieces of DNA from different genomes using enzymes. The latter two events involved the development of techniques used to transfer foreign DNA into new host cells.
Discovering the Cut-and-Paste Enzymes
The first major step forward in the ability to chemically modify genes occurred when American biologist Martin Gellert and his colleagues from the National Institutes of Health purified and characterized an enzyme in Escherichia coli responsible for the actual joining, or recombining, of separate pieces of DNA (Zimmerman et al., 1967). They called their find "DNA-joining enzyme," and this enzyme is now known as DNA ligase. All living cells use some version of DNA ligase to "glue together" short strands of DNA during replication. Using E. coli extract, the researchers next showed that only in the presence of ligase was it possible to repair single-stranded breaks in λ phage DNA. (Discovered in 1950 by American microbiologist Esther Lederberg, λ phage is a virus particle that infects E. coli.) More specifically, they showed that the enzyme was able to form a 3'-5'-phosphodiester bond between the 5'-phosphate end of the last nucleotide on one DNA fragment and the 3'-OH end of the last nucleotide on an adjacent fragment. The identification of DNA ligase was the first of several key steps that would eventually empower scientists to attempt their own recombination experiments—experiments that involved not just recombining the DNA of a single individual, but recombining DNA from different individuals, including different species.
A second major step forward in gene modification was the discovery of restriction enzymes, which cleave DNA at specific sequences. These enzymes were discovered at approximately the same time as the first DNA ligases by Swiss biologist Werner Arber and his colleagues while they were investigating a phenomenon called host-controlled restriction of bacteriophages. Bacteriophages are viruses that invade and often destroy their bacterial host cells; host-controlled restriction refers to the defense mechanisms that bacterial cells have evolved to deal with these invading viruses. Arber's team discovered that one such mechanism is enzymatic activity provided by the host cell. The team named the responsible enzymes "restriction enzymes" because of the way they restrict the growth of bacteriophages. These scientists were also the first to demonstrate that restriction enzymes damage invading bacteriophages by cleaving the phage DNA at very specific nucleotide sequences (now known as restriction sites). The identification and characterization of restriction enzymes gave biologists the means to cut specific pieces of DNA required (or desired) for subsequent recombination.
Inserting Foreign DNA into a New Host Cell
Although Griffith and Avery had had demonstrated the ability to transfer foreign genetic material into cells decades earlier, this "transformation" was very inefficient, and it involved "natural" rather than manipulated DNA. Only in the 1970s did scientists begin to use vectors to efficiently transfer genes into bacterial cells. The first such vectors were plasmids, or small DNA molecules that live naturally inside bacterial cells and replicate separately from a bacterium's chromosomal DNA.
Plasmids' utility as a DNA shuttle, or vector, was discovered by Stanford University biochemist Stanley Cohen. Scientists had already established that some bacteria had what were known as antibiotic resistance factors, or R factor-plasmids that replicated independently inside the bacterial cell. But scientists knew little about how the different R factor genes functioned. Cohen thought that if there were an experimental system for transforming host bacterial cells with these R-factor DNA molecules, he and other researchers might be able to better understand R-factor biology and figure out exactly what it was about these plasmids that made bacteria antibiotic-resistant. He and his colleagues developed that system by demonstrating that calcium chloride-treated E. coli can be genetically transformed into antibiotic-resistant cells by the addition of purified plasmid DNA (in this case, purified R-factor DNA) to the bacteria during transformation (Cohen et al., 1972).
Recombinant Plasmids in Bacteria
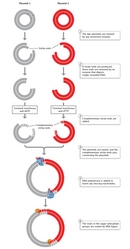
The following year, Stanley Cohen and his colleagues were also the first to construct a novel plasmid DNA from two separate plasmid species which, when introduced into E. coli, possessed all the nucleotide base sequences and functions of both parent plasmids. Cohen's team used restriction endonuclease enzymes to cleave the double-stranded DNA molecules of the two parent plasmids. The team next used DNA ligase to rejoin, or recombine, the DNA fragments from the two different plasmids (Figure 2). Finally, they introduced the newly recombined plasmid DNA into E. coli. The researchers were able to join two DNA fragments from completely different plasmids because, as they explained, "the nucleotide sequences cleaved are unique and self-complementary so that DNA fragments produced by one of these enzymes can associate by hydrogen-bonding with other fragments produced by the same enzyme" (Cohen et al., 1973).
The same could be said of any DNA—not just plasmids—from two different species. This universality—the capacity to mix and match DNA from different species, because DNA has the same structure and function in all species and because restriction and ligase enzymes cut and paste the same ways in different genomes—makes recombinant DNA biology possible.
Today, the E. coli λ bacteriophage is one of the most widely used vectors used to carry recombinant DNA into bacterial cells. This virus makes an excellent vector because about one-third of its genome is considered nonessential, meaning that it can be removed and replaced by foreign DNA (i.e., the DNA being inserted). As illustrated in Figure 3, the nonessential genes are removed by restriction enzymes (the specific restriction enzyme EcoRI is shown in the figure), the foreign DNA inserted in their place, and then the final recombinant DNA molecule is packaged into the virus's protein coat and prepped for introduction into its host cell.
Vectors Used in Mammalian Cells
A fourth major step forward in the field of recombinant DNA technology was the discovery of a vector for efficiently introducing genes into mammalian cells. Specifically, researchers learned that recombinant DNA could be introduced into the SV40 virus, a pathogen that infects both monkeys and humans. Indeed, in 1972, Stanford University researcher Paul Berg and his colleagues integrated segments of λ phage DNA, as well as a segment of E. coli DNA containing the galactose operon, into the SV40 genome. (The E. coli galactose operon is a cluster of genes that plays a role in galactose sugar metabolism.) The significance of their achievement was its demonstration that recombinant DNA technologies could be applied to essentially any DNA sequences, no matter how distantly related their species of origin. In their words, these researchers "developed biochemical techniques that are generally applicable for joining covalently any two DNA molecules" (Jackson et al., 1972). While the scientists didn't actually introduce foreign DNA into a mammalian cell in this experiment, they provided (proved) the means to do so.
Recombinant DNA Technology Creates Recombinant Animals
The first actual recombinant animal cells weren't developed until about a decade after the research conducted by Berg's team, and most of the early studies involved mouse cells. In 1981, for example, Franklin Costantini and Elizabeth Lacy of the University of Oxford introduced rabbit DNA fragments containing the adult beta globin gene into murine (mouse) germ-line cells (Costantini & Lacy, 1981). (The beta globins are a family of polypeptides that serve as the subunits of hemoglobin molecules.) Another group of scientists had demonstrated that foreign genes could be successfully integrated into murine somatic cells, but this was the first demonstration of their integration into germ cells. In other words, Costantini and Lacy were the first to engineer an entire recombinant animal (albeit with relatively low efficiency).
Interestingly, not long after the publication of his team's 1972 study, Paul Berg led a voluntary moratorium in the scientific community against certain types of recombinant DNA research. Clearly, scientists have always been aware that the ability to manipulate the genome and mix and match genes from different organisms, even different species, raises immediate and serious questions about the potential hazards and risks of doing so—implications still being debated today.
Since these early studies, scientists have used recombinant DNA technologies to create many different types of recombinant animals, both for scientific study and for the profitable manufacturing of human proteins. For instance, mice, goats, and cows have all been engineered to create medically valuable proteins in their milk; moreover, hormones that were once isolated only in small amounts from human cadavers can now be mass-produced by genetically engineered cells. In fact, the entire biotechnology industry is based upon the ability to add new genes to cells, plants, and animals As scientists discover important new proteins and genes, these technologies will continue to form the foundation of future generations of discoveries and medical advances.
References and Recommended Reading
Cohen, S. N., et al. Nonchromosomal antibiotic resistance in bacteria: Genetic transformation of Escherichia coli by R-factor DNA. Proceedings of the National Academy of Sciences 69, 2110–2114 (1972)
———. Construction of biologically functional bacterial plasmids in vitro. Proceedings of the National Academy of Sciences 70, 3240–3244 (1973)
Costantini, F., & Lacy, E. Introduction
of a rabbit beta-globin gene into the mouse germ line. Nature 294, 92–94 (1981) (link to article)
Crea, R., et al. Chemical synthesis of genes for human insulin. Proceedings of the National Academy of Sciences 75, 5765–5769 (1978)
GloFish. GloFish home page. www.glofish.com (Accessed July 3, 2008)
Jackson, D. A., et al. Biochemical method for inserting new genetic information into DNA of simian virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proceedings of the National Academy of Sciences 69, 2904–2909 (1972)
Kiermer, V. The dawn of recombinant DNA. Nature Milestones: DNA Technologies, http://www.nature.com/milestones/miledna/full/miledna02.html (2007) (link to article)
Miller, H. I. FDA on transgenic animals—A dog's breakfast? Nature Biotechnology 26, 159–160 (2008) (link to article)
Zimmerman, S. B., et al. Enzymatic joining of DNA strands: A novel reaction of diphosphopyridine nucleotide. Proceedings of the National Academy of Sciences 57, 1841–1848 (1967)










