



















« Prev Next »
Variation in Population Divergence Among Genomic Regions
Independent evolution of populations leads to their genetic divergence. Different regions of the genome are expected to exhibit highly variable levels of genetic divergence (reviewed by Nosil et al. 2009), ranging from genomic regions exhibiting little to no differentiation between populations to regions where genetic divergence is extremely pronounced (Figure 1). This pattern of variation in population divergence across regions of the genome has been referred to as heterogeneous genomic divergence (Nosil et al. 2009). Genomic divergence is expected to be highly heterogeneous during the process of population divergence and species formation, during which genetic differentiation associated with divergent natural selection accumulates in some regions while the homogenizing effects of gene flow or inadequate time for random differentiation by genetic drift preclude divergence in other regions. Many factors contribute to heterogeneous genomic divergence, including selection arising from ecological causes or genetic conflict, the stochastic effects of genetic drift, variable mutation rates, the genetic basis of traits under selection, and genetic linkage among genes on chromosomes. The patterns of genomic differentiation among populations record and integrate across these different historical evolutionary and genetic processes, thereby offering biologists opportunities to reconstruct the forces that shaped evolutionary divergence.

Figure 1: Three arbitrary classes of loci embedded within a continuous distribution of FST values
(A) Selected loci, or those very tightly linked to them, exhibit particularly high FST and thus are recognized as unlikely, statistical outliers. (B) Neutral loci that are weakly-linked to selected loci exhibit FST values that are somewhat elevated, but insufficiently so to be designated outlier loci. (C) Neutral unlinked loci exhibit low FST.
© 2010 Nature Education Modified from Egan et al. (2008) and reprinted with permission of Wiley-Blackwell. All rights reserved. 
We focus here on the contributions of divergent natural selection to population divergence. Such divergent selection will pull apart allele frequencies between populations at loci under selection and those physically-linked to them, resulting in strong differentiation of regions affected by selection. This might occur even if the remainder of the genome remains relatively undifferentiated. Thus, loci under divergent selection and those tightly physically-linked to them should exhibit greater differentiation than weakly-linked or unlinked neutral regions. If only a subset of loci experiences divergent selection, the selected loci can be recognized as exceptional relative to the remainder of the genome and as outlier loci whose genetic divergence exceeds neutral expectations (Figure 2).
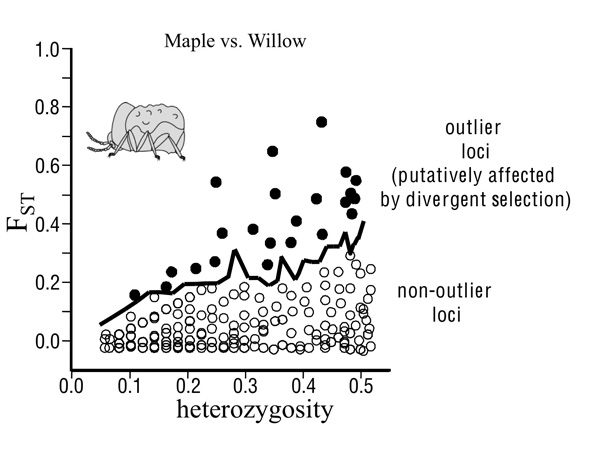
Figure 2: Heterogeneous genomic divergence in a genome scan of a pair of Neochlamisus bebbianae leaf beetle populations
Simulations determine the upper level of genetic divergence expected under neutral divergence due to genetic drift in the pair of populations, and loci that exceed this neutrality threshold (solid line, in this case 95% quantile) are recognized as outlier loci that are likely to have evolved under divergent selection.
© 2010 Nature Education Modified from Egan et al. (2008) and reprinted with permission of Wiley-Blackwell. All rights reserved.
The field of population genomics examines patterns in the genome, often using genome scans that examine genetic divergence between populations at numerous loci across the genome. The degree of genetic divergence is often measured using fixation indices such as FST, with larger index values representing greater differentiation between populations.
A Brief History of Population Genomics
Population genomic analyses require multi-locus data sets from multiple populations and identify non-neutral or outlier loci by contrasting patterns of population divergence among genetic regions. This approach was first proposed by Lewontin & Krakauer (1973), and numerous variations on this original method now exist (Beaumont 2005, Foll & Gaggiotti 2008, Gompert et al. [in review]). Perhaps the most commonly employed of these methods, particularly in non-model organisms, is the FST outlier analysis developed by Beaumont & Nichols (1996). This test contrasts FST for individual loci with an expected null distribution of FST based on a neutral model. Under this approach, loci with very high levels of differentiation between populations (i.e., high FST) are considered candidates for positive or divergent selection whereas loci with exceptionally low FST are regarded as candidates for balancing selection. However, many FST outlier analyses may be biased by departures from the assumed demographic history (Excoffier et al. 2009). An alternative approach to obtain a null distribution of population genetic differentiation is to assume that FST for individual loci represent independent draws from a common, underlying distribution that characterizes the neutral divergence across the genome and can be estimated directly from multi-locus data (Foll & Gaggiotti 2008; Figure 3). This alternative approach is more robust to different demographic histories. Recent advances in computational methods and molecular biology (including next-generation sequencing) allow patterns of genomic divergence to be investigated at previously unattainable scales. We now turn to describing in some more detail three case studies of population genomic analyses.

Figure 3: Statistics of population differentiation can be estimated by Bayesian methods, which provide a probabilistic framework in which to interpret differentiation of individual loci.
Differentiation among populations of Lycaeides butterflies in North America was estimated (Gompert et al. 2010) and summarized in a genome-level estimate of ΦST (a) and its average deviation (b). This analysis illustrated the high variation among loci across the genome (c, arbitrary set of 200 scored loci), some of which is likely to have been shaped by divergent natural selection.
© 2010 Nature Education All rights reserved.
Case Studies
Population Genomic Evidence for Recent and Rapid Evolutionary Adaptation in Humans
As humans expanded into new geographic regions and came to occupy novel environments, our ancestors experienced diverse patterns of natural selection that were recorded in the genomes of divergent populations. For example, humans moved from low elevations to occupy some of the highest plateaus and mountain ranges in the world, including the plateaus of Central Asia and the Andes of South America. These populations exhibit heritable physiological attributes that allow individuals to function at high altitudes (3250–4500 m) with low oxygen concentrations that are challenging to humans from lower elevations. Interestingly, humans from the Tibetan plateau exhibit several physiological attributes that differ from those of Andean highlanders, suggesting that independent evolutionary trajectories led to different adaptations to high altitude. Three studies have used genome scans to identify genes with exceptional allele frequency shifts between populations that were likely targets of divergent natural selection in Tibetan highlanders relative to other human populations (reviewed in Storz 2010). Several genes were identified as associated with a history of positive selection in Tibetan highlanders; the gene EPAS1 was identified as one of the most exceptional genes in each study and was also shown to be associated with presumably adaptive variation in hemoglobin concentration.
A second example of adaptive divergence among human populations is related to the persistence of lactase production in adults. Lactase production in the gut functions in the digestion of the milk sugar lactose, and lactase production in adults is prevalent in humans with ancestry in northern and western Europe and pastoralist populations in several regions of the world. Adult persistence of lactase is much less common in southern Europe and the Middle East and rare in non-pastoralist populations in Asia and Africa. Genetic studies have associated adult persistence of lactase with different genes in different populations that exhibit the trait at high frequency, indicating that the trait has arisen independently in multiple populations (Tishkoff et al. 2007). Remarkably, genomic variation surrounding each of the underlying genes is consistent with strong natural selection within the last several thousand years increasing the frequency of the derived, adaptive alleles (Tishkoff et al. 2007).
Genomic Islands in Anopheles Mosquitoes
To help understand regions of divergence in the genome, evolutionary biologists have developed the concept of "genomic islands of divergence" (Turner et al. 2005; Figure 4). A genomic island is any gene region, be it a single nucleotide or an entire chromosome, that exhibits significantly greater differentiation than expected under neutrality (and thus generally greater divergence than observed in neighboring genomic regions). The metaphor, therefore, draws a parallel between genetic differentiation observed along a chromosome and the topography of oceanic islands and the contiguous sea floor to which they are connected. Following this metaphor, sea level represents the threshold above which observed differentiation is significantly greater than expected by neutral evolution alone. Thus, an island is composed of both directly selected and tightly linked (potentially neutral) loci.
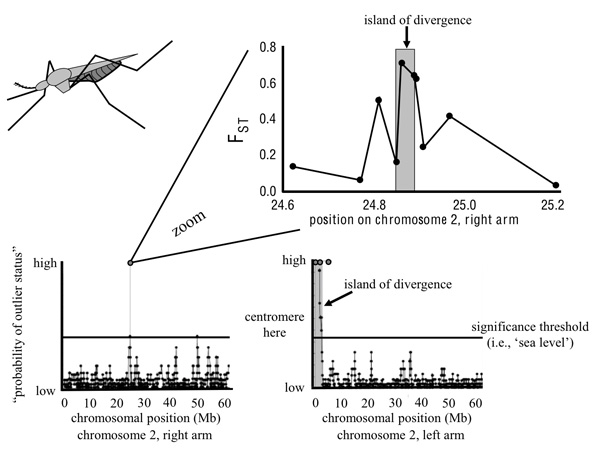
Figure 4: An empirical example of genomic islands of divergence involving incipient species of Anopheles gambiae
The bottom two panels depict patterns of differentiation across chromosome two (Turner et al. 2005). Grey areas were identified as highly differentiated in sliding window analyses, with differentiation further confirmed by sequencing loci within these regions (red circles). A large island of exceptionally high differentiation is evident on the left arm, near the centromere. A small island is also evident on the right arm. The top panel depicts a subsequent study (Turner & Hahn 2007) where portions of all annotated genes within the smaller island were sequenced.
© 2010 Nature Education Modified from the original studies and reprinted with permission of the Public Library of Science and the Society of Molecular Biology and Evolution. All rights reserved.
Figure 4 depicts an empirical example of a genomic island from population genomic studies of different forms of Anopheles gambiae mosquitoes. These insects are vectors for malaria, and Turner et al. (2005) surveyed divergence across the genome of the different mosquito forms. They found just a few regions that were differentiated (i.e., a few isolated genomic islands) between the forms. In a follow-up study, Turner & Hahn (2007) sequenced portions of all annotated genes within one of the islands. As expected, sequence differentiation peaked within the "island" but the fine-scale data allowed more detailed characterization of the nature of the island, showing, for example, that differentiation drops off rapidly with distance from the region of maximum differentiation.
Other Applications of Population Genomics
In addition to genome scans for differentiation, population genomics also includes many other diverse analyses of population genomic variation. These include research that more directly ties or genetically maps variation in phenotypes (e.g., body size, bill length, feather color, mating behavior) to genetic variation in various organisms. Similarly, researchers use other signatures of natural selection (e.g., extended haplotype blocks) to detect genomic regions that are likely to have experienced selection. As technology has allowed the rate of genomic data acquisition to increase enormously, an increasing range of biological questions can now be addressed at the scale of the genome rather than focusing on very small fractions of genomic variance.
Caveats
Genome scans of differentiation, by virtue of detecting divergent selection via looking for the most differentiated regions, are inevitably destined to underestimate how widespread the effects of selection are in the genome. In other words, genome scans will often fail to identify regions that are affected by divergent selection but only weakly so. A recent analysis of divergent selection in two host forms of Rhagoletis flies exemplifies this issue (Michel et al. 2010). This study reported that standard outlier analyses detected evidence for selection on only a few exceptionally differentiated genomic regions. In contrast, results from an experiment where the fly's genome was directly subjected to divergent selection revealed that selection was affecting much of the genome, albeit often quite weakly. The prevalence of selection may also be underestimated because most analytical methods are unlikely to detect soft sweeps involving smaller shifts in the allele frequency spectrum of multiple loci and are dependent on the genetic architecture of adaptation.
Conclusions and Future Directions
Population genomics holds great promise for understanding the evolutionary processes affecting genomes. However, population genomics is not a panacea, and analyses must increasingly be conducted with care. For example, the stochastic nature of next-generation sequencing technologies creates uneven coverage among individuals and genetic regions, which misses data for many individuals and loci, and thus increased uncertainty in the genotypes of individuals relative to traditional Sanger sequencing. Appropriately modeling and accounting for this uncertainty is important and preferable to discarding large amounts of sequence data (Gompert et al. 2010). Further advances in molecular and computational biology and increased computing power itself will allow more powerful and accurate application of population genomics.
References and Recommended Reading
Beaumont, M. A. Adaptation and speciation: what can F-st tell us? Trends in Ecology and Evolution 20, 435–440 (2005).
Beaumont, M. A. & Nichols, R. A. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London B 263, 1619–1626 (1996).
Egan, S. P. et al. Selection and genomic differentiation during ecological speciation: isolating the contributions of host-association via a comparative genome scan of Neochlamisus bebbianae leaf beetles. Evolution 62, 1162–1181(2008).
Excoffier, L. et al. Detecting loci under selection in a hierarchically structured population. Heredity 103, 285–298 (2009).
Foll, M. & Gaggiotti, O. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Gompert, Z. et al. Bayesian analysis of molecular variance in pyrosequences quantifies population genetic structure across the genome of Lycaeides butterflies. Molecular Ecology 19, 2455–2473 (2010).
Gompert Z. & Buerkle C. A. Analytical tools for next-generation sequence data. In review.
Lewontin, R. C. & Krakauer, J. Distribution of gene frequency as a test of the theory of selective neutrality of polymorphisms. Genetics 74, 175–195 (1973).
Michel, A. P. et al. Widespread genomic divergence during sympatric speciation. Proceedings of the National Academy of Sciences USA 107, 9724–9729 (2010).
Nosil, P. et al. Divergent selection and heterogeneous genomic divergence. Molecular Ecology 18, 375–402 (2009).
Storz, J. F. Genes for high altitudes. Science 329, 40–41 (2010).
Tishkoff, S. A. et al. Convergent adaptation of human lactase persistence in Africa and in Europe. Nature Genetics 39, 31–40 (2007).
Turner, T. L., & Hahn, M. W. Locus- and population specific selection and differentiation between incipient species of Anopholes gambiae. Molecular Biology and Evolution 24, 2132–2138 (2007).
Turner, T. L. et al. Genomic islands of speciation in Anopheles gambiae. Public Library of Science Biology 3, 1572–1578 (2005).










