
Abstract
The view of the nervous system being the victim of destructive inflammation during autoimmunity, degeneration, or injury has been rapidly changing. Recent studies are supporting the idea that the immune system provides support for the nervous system at various levels. Though cell patrolling through the nervous system parenchyma is limited compared with other tissues, immune cell presence within the central nervous system (CNS; microglia), as well as around it (in the meningeal spaces and choroid plexus) has been shown to be important for brain tissue maintenance and function. This review primarily explores recent findings concerning neuroimmune interactions and their mechanisms under homeostatic conditions.
Similar content being viewed by others
INTRODUCTION
The immune system, although long known for its essential function in the organism’s defense against pathogens, has recently also become appreciated as indispensable for tissue maintenance. In addition to constant patrolling by circulating immune cells, every organ, including the brain, benefits from a dedicated resident immune system. Under pathological conditions, both the tissue-resident and the peripheral immunity react rapidly to eliminate the threat and facilitate tissue recovery. The central nervous system (CNS) does not seem to be much different in this regard, despite it long being regarded as an ‘ivory tower,’ immune-privileged organ. Under physiological conditions, although immune access to the CNS is indeed tightly regulated, the system nevertheless derives essential benefit from parenchymal resident microglia, as well as from an immune repertoire in the meninges and choroid plexus. In this review we discuss the different levels of immune support for the CNS and consider how the nervous system is affected by immune system function in health and disease.
THE BRAIN-RESIDENT IMMUNE SYSTEM
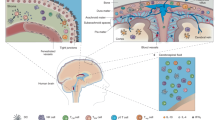
The parenchymal tissue of the CNS contains a limited immune repertoire, composed mainly of resident microglia and perivascular macrophages (although the perivascular space is external to the parenchyma, leaving microglia as the main true immune cell within this part of the CNS) (Figure 1). In the course of embryonic development, the brain is seeded by microglia derived from primitive yolk-sac macrophages (Ginhoux et al, 2010; Gomez Perdiguero et al, 2015; Sheng et al, 2015). During normal postembryonic homeostasis the microglia proliferate within the parenchyma, with no infiltration from the periphery (Ajami et al, 2011; Gomez Perdiguero et al, 2015; Mildner et al, 2007). Although the presence and many macroscopic/histological features of microglia have been known for almost a century (Kettenmann et al, 2011), understanding their full range of functions has proved to be challenging. Microglia were first described by del Rio Hortega, who went on to advance the idea that microglia in the CNS behave like microphages in other sites of the body (Kettenmann et al, 2011). Modern RNA-seq analyses show that the microglial transcriptome shares homology with tissue-resident macrophages (Gautier et al, 2012) while also exhibiting their unique genetic signature. For example, microglia respond rapidly during infection and injury, adopting the characteristic amoeboid morphology (Davalos et al, 2005; Roth et al, 2014). When this happens, microglia increase their migratory and phagocytic programs (Fourgeaud et al, 2016; Roth et al, 2014) and become phenotypically indistinguishable from macrophages (Mildner et al, 2007; Yamasaki et al, 2014).

Layers of immune surveillance of the central nervous system (vs peripheral organs). The nervous system has long been viewed as isolated from the immune system because of the lack of circulating immune cell infiltration during homeostatic conditions. However, we have recently begun to appreciate that the nervous system benefits from extensive immune support, as well as extensive regulatory mechanisms. The immune presence in the parenchyma (constituted by microglia) is limited, with compartmentalization of a full complement of immune cells in the meninges (and partial in the choroid plexus). Responses from the peripheral immunity translate into immune responses in the meninges and choroid plexus that may then be relayed, in an attenuated manner, to the parenchyma. Such layering allows for protection of the CNS from destructive inflammation (which can be devastating for a bone-enclosed organ sensitive to mechanical stimuli) while still being able to receive, and thus respond to, environmental stimuli. Peripheral organs (exemplified here by the liver) also benefit from specialized tissue-resident immune cells (ie, Kupffer cells in the liver). However, most organs do not benefit from the extra layers of checkpoint (such as meninges in the CNS) for entry of immune cells, allowing them to readily infiltrate directly from the circulation.
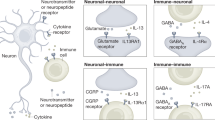
Less clear are the roles of microglia in the absence of infectious or injury stimuli. With the advancement of in vivo imaging techniques, the scientific community began to appreciate the dynamic nature of microglia during their resting steady state (Davalos et al, 2005; Nimmerjahn et al, 2005; Tremblay et al, 2010; Wake et al, 2009). These cells constantly sample their environment by extending and retracting their processes, and can be observed making contacts with synaptic formations (Paolicelli et al, 2011; Tremblay et al, 2010; Wake et al, 2009). The synaptic contacts appear to be mediated by soluble factors such as ATP and soluble fractalkine, whose release can be mediated by neuronal activity (Dissing-Olesen et al, 2014; Eyo et al, 2014; Fontainhas et al, 2011; Haynes et al, 2006; Hoshiko et al, 2012; Li et al, 2012; Paolicelli et al, 2011; Sipe et al, 2016). Both raised and lowered neuronal activity seems to trigger synaptic contacts by microglia, although the microglial behavior subsequent to such contact may be different in each case. Glutamate stimulation of either NMDA or AMPA receptors, as well as evoked circuit activity, can drive the release of ATP, inducing elongation of microglial processes via activation of the P2Y12 purinergic receptors on microglia (Dissing-Olesen et al, 2014; Eyo et al, 2014; Fontainhas et al, 2011; Haynes et al, 2006; Li et al, 2012). The functions of microglial contacts under these circumstances are largely unknown. In a seizure model, loss of signaling through P2Y12 prevents activity-driven extension of microglia and leads to exacerbated seizure severity (Eyo et al, 2014), indicating that microglia might play a role in glutamate buffering during intense neuronal activity. On the other hand, decreased neuronal activity, for example by sensory deprivation or inhibition of excitatory signaling, can also increase synaptic contacts by microglia in an ATP-dependent manner (Sipe et al, 2016; Tremblay et al, 2010). In such cases, the decrease in neuronal activity causes extension of processes but arrest of microglial motility, leading ultimately to an increased microglial intake of synaptic elements (Sipe et al, 2016; Tremblay et al, 2010).
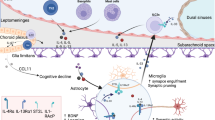
Synaptic pruning by microglia is important for both normal development and maintenance of homeostasis in adulthood. Upon extension of microglial processes, microglial receptors such as CX3CR1, CR3, CD200R, or TREM2/DAP12 may recognize ligands on the neuronal membrane (Hoshiko et al, 2012; Paolicelli et al, 2011; Poliani et al, 2015; Schafer et al, 2012). Perhaps the oldest documented interaction is that between fractalkine (on neurons) and CX3CR1 (on microglia). Mice deficient in CX3CR1 signaling exhibit deficits in the numbers of microglia during development, as well as decreased synaptic pruning (Paolicelli et al, 2011). Although the microglia in these mice reach normal numbers in adulthood, they appear to have an exuberant production of inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα) (Cardona et al, 2006; Cho et al, 2011; Dudley et al, 2002). Moreover, the mice maintain deficits in synaptic maturation (Hoshiko et al, 2012) and function (Dudley et al, 2002; Zhang et al, 2014) that translate behaviorally into cognitive and social impairments (Cho et al, 2011; Dudley et al, 2002; Zhang et al, 2014). It is important to note that although many of these effects may be a result of deficient synaptic pruning, they can also be ascribed to a microglial effect on neuronal circuit activity via other mechanisms in response to soluble CX3CL1 (Ragozzino et al, 2006).
A more recently described pathway for microglial pruning is the complement system (Stevens et al, 2007), in which weaker synaptic inputs on developing neurons are decorated with C1q and C3 molecules capable of engaging CR3 receptors on microglia for targeted elimination (Schafer et al, 2012; Stevens et al, 2007). Classical and nonclassical signaling of complement components is also active in adulthood and aging, but its implications for microglia are unknown. In aged mice, for example, C1q is increased both at synapses and on microglia, and C1q- and C3-knockout mice are protected from cognitive impairments (Shi et al, 2015; Stephan et al, 2013). Increased complement decoration at synapses can be induced by oligomeric Aβ, and is also observed in mouse models of Alzheimer’s disease. In this context, it appears that there is excessive microglial engulfment of synapses in a CR3-dependent manner (Hong et al, 2016).
Although the evidence described above points to important roles for microglia in wiring of the brain, more recent efforts have been directed toward understanding the roles of microglia specifically in adulthood. Studies have employed both genetic (CX3CR1CreER/+:R26iDTR/+) and pharmacological approaches (CSF1 antagonists such as PLX3397) to cause extensive microglial death in the adult or aged CNS (Elmore et al, 2014; Parkhurst et al, 2013). The short-term effects of such drastic approaches appear to be subtle, expressed as mild cognitive impairments (Parkhurst et al, 2013) or with no behavioral effects at all (Elmore et al, 2014). Ablation of microglia does not induce neuronal loss, but longer-term ablation can cause synaptic degradation (Parkhurst et al, 2013; Spangenberg et al, 2016; Wang et al, 2016). Intriguingly, in mouse models of Alzheimer’s disease with its hallmark microglial activation and proliferation, the effects of microglial ablation are beneficial (Hong et al, 2016; Olmos-Alonso et al, 2016; Spangenberg et al, 2016). In particular, loss of microglia prevents spine loss and cognitive impairments, but has no effect on Aβ levels.
Taken together, recent evidence points to discrete roles for microglia in circuit wiring, aiding neuronal function, and maintaining CNS homeostasis. However, under conditions of neuronal degradation and low-grade inflammation (such as cognitive decline and aging), microglia appear to accelerate both the functional loss of neurons and the cognitive decline.
CHOROID PLEXUS: AN ENTRY POINT FOR PATROLLING IMMUNE CELLS?
The main function of the choroid plexus, an epithelial tissue located within the ventricles of the brain, is filtration of the blood to yield the cell-free low-protein cerebrospinal fluid (CSF). Although the cell count in the CSF is low, it is not negligible (1000−3000 cells per ml in humans; Kivisakk et al, 2003), and an increase in the number is taken as a sign of neuroinflammation (Giunti et al, 2003). Immune cells are also found within the choroid plexus epithelium, and during inflammatory events their numbers increase (Giunti et al, 2003; Young et al, 2011), giving rise to the hypothesis that the choroid plexus is one of the points of immune-cell entry into the CSF. Adhesion molecules, such as intercellular adhesion molecule 1 (ICAM1) and P-selectin, are expressed in the choroid plexus at baseline, providing a permissive environment for immune-cell infiltration (Baruch et al, 2015; Kivisakk et al, 2003). The levels of such adhesion molecules, as well as of tight junction components, are dynamically controlled by the cytokine milieu and inflammatory stimuli to regulate the amounts of cell traffic into the CSF (Baruch et al, 2015; Kunis et al, 2013; Marques et al, 2009; Zhang et al, 2013). Inflammatory stimuli, such as peripheral infection or autoimmune diseases, are usually ‘activators’ of the choroid plexus (Marques et al, 2007, 2009; Petito and Adkins, 2005; Reboldi et al, 2009; Shrestha et al, 2014; Young et al, 2011; Zhang et al, 2013). In contrast, an aging milieu renders the site more quiescent for immune-cell trafficking (Baruch et al, 2013, 2015; Mesquita et al, 2015).
‘Activation’ of the choroid plexus usually refers to upregulation of inflammatory cytokines (such as IL-1β, TNFα, and IL-6), as well as upregulation of adhesion molecules and downregulation of tight junction molecules (Marques et al, 2007, 2009; Shrestha et al, 2014; Young et al, 2011). Interestingly, the inflammatory response to peripheral stimuli in the choroid plexus is much weaker than in other organs in the periphery (liver, spleen) and is subjected to tight temporal regulation, usually resolving within 24 h of the stimulus (Marques et al, 2009; Shrestha et al, 2014). As a result, peripheral stimuli in the choroid plexus normally dictate the entry of only a limited, nonantigen-specific immune-cell infiltrate into the CSF (Petito and Adkins, 2005; Shrestha et al, 2014; Young et al, 2011). Under autoimmune conditions, however, entry of the infiltrate is more extensive (Giunti et al, 2003; Shrestha et al, 2014). In addition to the temporal regulation (whose mechanisms are as yet unknown), other mechanisms, such as shedding of syndecans, may limit the extent of the choroid plexus infiltrate (Zhang et al, 2013). During autoimmune encephalitis, syndecan is shed from the choroid plexus epithelium into the CSF, where it acts like a sponge to bind released chemokines, thus sequestering them from accumulating lymphocytes (Zhang et al, 2013).
More recent work has focused on the role of the choroid plexus as the site for neuroimmune interactions in aging and models of Alzheimer’s disease. Lymphocyte levels in CSF samples from aged individuals are decreased, whereas tau protein accumulates and monomeric Aβ decreases (Lueg et al, 2015). In a mouse model of Alzheimer’s disease, Aβ protein accumulates in the CSF much earlier (after 3 months) than its deposition is detectable in the brain parenchyma (after 12 months) (Mesquita et al, 2015). Moreover, studies in aged mice showed that the cytokine milieu of the choroid plexus is shifted toward a type 2 skew, with increased IL-4 and decreased interferon-γ (IFNγ) levels (Baruch et al, 2013; Mesquita et al, 2015). At the same time, mice with impaired IFNγ signaling show a decrease in adhesion molecules in the choroid plexus and impaired immune-cell trafficking at this site, indicating a possible role for type 1 signaling in maintaining immune function in the CNS (Kunis et al, 2013; Raposo et al, 2014). Transient suppression of regulatory T cells in mouse model of Alzheimer’s disease resulted in upregulation of adhesion molecules in the choroid plexus, as well as increased lymphocyte recruitment (Baruch et al, 2015). These mice also exhibited improved cognitive performance, indicating that increased immune activity at the choroid plexus is beneficial in this model.
Studies have indicated that under pathological conditions the choroid plexus can be an active site for immune trafficking into the CNS, but they do not exclude other routes, such as through the meningeal vasculature (Young et al, 2011). A new study, using rats with experimental autoimmune encephalomyelitis (EAE) as a model, indeed points to the meningeal blood vessels as the main site for immune-cell trafficking at specific time points during EAE development (Schlager et al, 2016). One possibility is that at certain time points during the inflammatory process, infiltration through the choroid plexus is regulated via specific pathways (such as those mediated by CCR6 or syndecan) (Reboldi et al, 2009; Zhang et al, 2013). It seems, however, that more thorough characterization of the immune responses in each compartment of the CNS is needed. One question that the field is actively trying to address is whether and how immune cells can infiltrate into the parenchyma once in the CSF. Though CSF washes throughout the brain, it is mostly confined to the ventricles and meningeal spaces. The recently described glymphatic system (Iliff et al, 2013; Iliff and Nedergaard, 2013) proposes that CSF can also drain along large arteries and from there into the interstitial space and back in the perivenular space for molecular clearance. Whether cells can also use this route to move from CSF into the parenchyma remains unknown. Alternatively, cells could transverse the pia mater or ventricular lining to enter the parenchyma. Regardless of the route, the cells would have both extracellular matrix and astrocyte barriers to overcome and direct evidence (ie, imaging) for either process is still lacking.
Also missing is knowledge of the homeostatic regulation of cell trafficking through the choroid plexus, as well as a better understanding of the function of immune cells in the CSF. Are they regulated by neuronal activity? Are they needed for immunosurveillance, or perhaps for secretion of tonic levels of cytokines? Do they sample CNS antigens to maintain tolerance? Addressing these questions is made more difficult by the lack of means to inhibit trafficking specifically through the choroid plexus.
MENINGEAL SPACES: IMMUNE SURVEILLANCE AROUND THE BRAIN
The meningeal compartment of the CNS contains a wide repertoire of immune cells within the membranes that surround the parenchymal tissue, rendering this compartment an immunologically competent site (Figure 1) (Bartholomaus et al, 2009; Derecki et al, 2010; Hatfield and Brown, 2015; Kim et al, 2009; Levy et al, 2007; Louveau et al, 2015; Sayed et al, 2010). Studies have shown that under inflammatory conditions the numbers of immune cells in the meningeal compartment can increase, possibly on a larger scale than in the choroid plexus (Bartholomaus et al, 2009; Kim et al, 2009; Kivisakk et al, 2009; Sayed et al, 2010; Schlager et al, 2016). For example, synchronous extravasion of neutrophil waves was observed in the meninges of murine viral meningitis models (Kim et al, 2009). Extensive infiltration in the meninges was also observed in rodents with EAE (Bartholomaus et al, 2009; Kivisakk et al, 2009; Schlager et al, 2016). Recent evidence suggests that such infiltrating autoimmune cells can then invade the parenchyma via the pial membrane (Schlager et al, 2016). In addition to the various molecular players that regulate infiltration, meningeal mast cells seem to play an important role in gatekeeping of immune cell infiltration during EAE, as well as in other inflammatory contexts, such as stroke (Arac et al, 2014; Christy et al, 2013; Sayed et al, 2010).
Though much less intensively investigated, meningeal immune responses during normal physiological conditions have also been reported, such as during learning (Derecki et al, 2010; Filiano et al, 2016). Following a learning task, immune cells accumulate in the meninges, exhibiting primarily a type-2 phenotype (Derecki et al, 2010, 2011). Unlike in severe inflammatory conditions, however, this response does not result in parenchymal infiltration. Older studies, showing meningeal mast cell degranulation after neuronal stimulation, suggest that neuronal activity can be one of drivers of meningeal immune responses (Dimitriadou et al, 1991). On the other hand, there is evidence that meningeal immune responses, such as degranulation of mast cells, can cause neuronal activation in the trigeminal nucleus via meningeal fibers (Chen et al, 2014; Karatas et al, 2013; Levy et al, 2007). Such studies render the meningeal spaces an exciting site for neuroimmune interactions and invite further exploration. We have yet to understand, for example, how meningeal immune responses influence neuronal processes and whether, in the absence of rampant inflammation, they translate into microglial responses. Recent characterization of the meningeal lymphatic vessels provides an exit route for immune cells and CNS antigens, enhancing our understanding of the complexity and regulation of immune responses in the CNS (Aspelund et al, 2015; Louveau et al, 2015). Recent works describe a previously unappreciated concentration of immune cells along the venous sinuses (Louveau et al, 2015), the origin or activity of which is yet unexplored. Given the observed accumulation of amyloid and tau proteins in the CSF long before their build-up in the parenchyma, are there dysregulated drainage and/or immune responses that may ultimately lead to Alzheimer’s disease pathology? Finding answers to such questions may change how we approach the understanding and treatment of neurodegeneration. Although tools for the study of local responses are limited, methods such as the novel application of pharmaceutical modulators of cell trafficking and activation through the thinned skull will facilitate the exploration of immune responses specifically at this site without causing damage to the CNS or widespread peripheral effects (Roth et al, 2014).
PERIPHERAL IMMUNITY: RIPPLES OF IMMUNE RESPONSES ON BRAIN HOMEOSTASIS AND FUNCTION
The ability of peripheral immune responses to affect brain function has been increasingly recognized over the past two decades (Dantzer et al, 2008; Rook et al, 2011; Yirmiya and Goshen, 2011). Perhaps the best documented example is that of sickness behavior, when a peripheral infection or an infection mimic (such as lipopolysaccharide (LPS)) causes a systemic immune response that, together with its accompanying storm of cytokines, affects brain function (Dantzer and Kelley, 2007; Kelley et al, 2003). Systemic administration of LPS has been shown to cause broad immune activation in the meninges and choroid plexus, as well as in the brain parenchyma (Chen et al, 2012; Gorina et al, 2011; Marques et al, 2009; Pascual et al, 2012; Zhang et al, 2014). Microglia and other glia in the brain respond to inflammatory stimuli by further secreting their own cytokines, thus propagating the immune response (Habbas et al, 2015; Marques et al, 2009; Zhang et al, 2014). Parenchymal increase of various inflammatory cytokines has detrimental effects on neuronal activity (such as decreased long-term potentiation) that translates to behavioral deficits in learning, in exploration, or in social interaction (Dantzer and Kelley, 2007; Kelley et al, 2003). Another example of immune activity affecting the brain is the maternal immune-activation model for autism, a model based on the strong association of autism diagnosis with maternal infection during gestation (Atladottir et al, 2010; Lee et al, 2015). In mice, this is contrived by injecting pregnant dams with the viral analog poly(I:C), thereby triggering an inflammatory response that is perpetuated in the pups (Choi et al, 2016; Hsiao et al, 2012; Smith et al, 2007). The mother’s transient immune response changes the development and responsiveness of the offspring’s immune system, rendering it more inflammatory (Choi et al, 2016; Hsiao et al, 2012; Smith et al, 2007). Overproduction of IL6 and high levels of Th17 cells following the immune response causes abnormal brain development, resulting in impaired neuronal migration and deficits in social interaction (Choi et al, 2016; Hsiao et al, 2012; Smith et al, 2007).
Although studies of inflammation and sickness behavior show that strong immune responses can be detrimental to CNS function, the same can be said of the lack of immune activity (Rook et al, 2011; Yirmiya and Goshen, 2011). A large body of literature has documented the finding that mice lacking an adaptive immune system (T and B lymphocytes) show impaired cognitive function and aberrant stress responses (Bartholomaus et al, 2009; Brynskikh et al, 2008; Derecki et al, 2010; Kipnis et al, 2004; Marques et al, 2009; Marsh et al, 2016; Ziv et al, 2006; Filiano et al, 2016). Such mice demonstrate spatial memory deficits and decreased adult neurogenesis (Derecki et al, 2010; Wolf et al, 2009; Ziv et al, 2006). Reconstitution of immunodeficient mice with a full complement of lymphocytes (through either bone marrow or adoptive transfer) restores their learning ability as well as their neurogenic capacity (Brynskikh et al, 2008; Derecki et al, 2010; Marsh et al, 2016). Interestingly, repopulation with CD4+ T cells alone (but not with CD8+ or B cells) is sufficient to rescue their phenotypes, indicating that helper T cells play an important role in supporting CNS functions (Wolf et al, 2009). We have yet to understand precisely how T-cell activity supports brain function, but certain mechanisms may prevent detrimental inflammatory responses in the meninges (Derecki et al, 2010, 2011). Intriguingly, recent evidence indicates that in the absence of autoimmune inflammation, T cells specific to CNS antigens may be of preferential importance for brain function (Baruch et al, 2013; Radjavi et al, 2014; Ziv et al, 2006). The role of the adaptive immune system in supporting brain function also becomes apparent during aging. In addition to a decrease in the number of newly generated lymphocytes, an accumulation of FOXP3+ regulatory T (Treg) cells is a hallmark of the aging immune system (Bapat et al, 2015; Montecino-Rodriguez et al, 2013; Raynor et al, 2015; van der Geest et al, 2014). Decreased immune activity and accumulation of regulatory T cells are reportedly associated not only with high tumor incidence but also with impaired cognition (Baruch et al, 2015; Koronyo et al, 2015; Marsh et al, 2016; Ron-Harel et al, 2008). Evidence suggests that a transient decrease in Treg cell numbers provides a boost in immune activity that benefits behavioral outcomes (Baruch et al, 2015; Ron-Harel et al, 2008). Although the mechanisms of this phenomenon have yet to be explored, boosting of the immune system may be an attractive option for improving cognitive function.
It should be noted that even though most of the above-cited studies have focused on peripheral immune responses, it is likely that these responses affect immune cells in the meninges and the choroid plexus as well (and possibly even microglia through soluble factors). Although the peripheral component may be sufficient to affect the brain via cytokine mediators acting through circumventricular organs, it is important to distinguish between the immune responses that occur at each site in order to identify the events that initiate them and develop specific treatments. As discussed in the previous section, local activation of mast cells in the meninges is necessary for immune infiltration during stroke (Arac et al, 2014). A recent study suggests that after stroke, Th17 cells can migrate from the intestines to the meninges to propagate neuroinflammation (Benakis et al, 2016). In this context, understanding the initiating events, timing, and recruitment sites could enable us to define more precise targets for limiting inflammation after stroke.
CONCLUSIONS
Over the past few decades it has become apparent that not only do the nervous and immune systems interact and influence each other, but also that such crosstalk takes place under homeostatic conditions. Given the parenchymal distribution of resident microglia and the wide repertoire of immune cells in the meninges, in many ways the brain is no different from other organs in terms of its immune presence and surveillance, although meningeal immunity represents an aspect of immune uniqueness of the CNS. Neuroscientists have only quite recently come to appreciate that CNS function is affected by immune activity from embryonic development to aging. We have learned, for example, that brain-resident microglia participate in the pruning and maintenance of neuronal circuits both early in life (during the wiring of the brain) and in adulthood (during processes of learning and refinement). We now also know that the activity of microglia, as well as of other cells of the nervous system, can be affected by signaling from the peripheral immune system. We are just beginning to appreciate the extent to which neuronal activity is influenced by immune system function, and is even dependent on it. Studies to date have shown that, as for peripheral organs, too much (sickness behavior) or too little (aging and immunodeficiency) immune activity is detrimental for CNS function; however, the regulatory mechanisms that balance neuroimmune interactions during homeostatic conditions have yet to be fully identified. We know that the immune system can signal the nervous system via cytokines, and that the nervous system can signal the immune system via innervation of lymphoid organs. We can also envision that ‘CNS-conditioned’ messages (delivered via cells, vesicles, or molecules) may be able to drain out of the nervous system and further orchestrate supportive immune responses.
The objective of this review was to highlight interactions between the immune and the nervous systems in the course of homeostatic processes such as learning or aging. Although studies addressing these topics are accumulating, the extent of neuroimmune interactions is still far from being understood. Fortunately, we now have more sensitive and a larger array of methods to track both immune and neuronal responses. Thus, we can look forward to the emergence of further studies in which the focus is shifted from models of inflammation to homeostatic processes.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011). Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14: 1142–1149.
Arac A, Grimbaldeston MA, Nepomuceno AR, Olayiwola O, Pereira MP, Nishiyama Y et al (2014). Evidence that meningeal mast cells can worsen stroke pathology in mice. Am J Pathol 184: 2493–2504.
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M et al (2015). A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212: 991–999.
Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M et al (2010). Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 40: 1423–1430.
Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A et al (2015). Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528: 137–141.
Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW et al (2009). Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 462: 94–98.
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W et al (2013). CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci USA 110: 2264–2269.
Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A et al (2015). Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun 6: 7967.
Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M et al (2016). Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med 22: 516–523.
Brynskikh A, Warren T, Zhu J, Kipnis J (2008). Adaptive immunity affects learning behavior in mice. Brain Behav Immun 22: 861–869.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9: 917–924.
Chen L, Li X, Huang L, Wu Q, Chen L, Wan Q (2014). Chemical stimulation of the intracranial dura activates NALP3 inflammasome in trigeminal ganglia neurons. Brain Res 1566: 1–11.
Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X et al (2012). Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32: 11706–11715.
Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P et al (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 286: 32713–32722.
Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV et al (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351: 933–939.
Christy AL, Walker ME, Hessner MJ, Brown MA (2013). Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE. J Autoimmun 42: 50–61.
Dantzer R, Kelley KW (2007). Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 21: 153–160.
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8: 752–758.
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR et al (2010). Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med 207: 1067–1080.
Derecki NC, Quinnies KM, Kipnis J (2011). Alternatively activated myeloid (M2) cells enhance cognitive function in immune compromised mice. Brain Behav Immun 25: 379–385.
Dimitriadou V, Buzzi MG, Moskowitz MA, Theoharides TC (1991). Trigeminal sensory fiber stimulation induces morphological changes reflecting secretion in rat dura mater mast cells. Neuroscience 44: 97–112.
Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA (2014). Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci 34: 10511–10527.
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ et al (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 19: 19.
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA et al (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82: 380–397.
Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ (2014). Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci 34: 10528–10540.
Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I et al (2016). Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 535: 425–429.
Fontainhas AM, Wang M, Liang KJ, Chen S, Mettu P, Damani M et al (2011). Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One 6: e15973.
Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG et al (2016). TAM receptors regulate multiple features of microglial physiology. Nature 532: 240–244.
Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S et al (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13: 1118–1128.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845.
Giunti D, Borsellino G, Benelli R, Marchese M, Capello E, Valle MT et al (2003). Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. J Leukoc Biol 73: 584–590.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L et al (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518: 547–551.
Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM (2011). Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59: 242–255.
Habbas S, Santello M, Becker D, Stubbe H, Zappia G, Liaudet N et al (2015). Neuroinflammatory TNFalpha impairs memory via astrocyte signaling. Cell 163: 1730–1741.
Hatfield JK, Brown MA (2015). Group 3 innate lymphoid cells accumulate and exhibit disease-induced activation in the meninges in EAE. Cell Immunol 297: 69–79.
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB et al (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9: 1512–1519.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S et al (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: 712–716.
Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci 32: 15106–15111.
Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH (2012). Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci USA 109: 12776–12781.
Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M et al (2013). Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123: 1299–1309.
Iliff JJ, Nedergaard M (2013). Is there a cerebral lymphatic system? Stroke 44 (6 Suppl 1): S93–S95.
Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD et al (2013). Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339: 1092–1095.
Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW. et al (2003). Cytokine-induced sickness behavior. Brain Behav Immun 17 (Suppl 1): S112–S118.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011). Physiology of microglia. Physiol Rev 91: 461–553.
Kim JV, Kang SS, Dustin ML, McGavern DB (2009). Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 457: 191–195.
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M (2004). T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA 101: 8180–8185.
Kivisakk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM et al (2009). Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol 65: 457–469.
Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T et al (2003). Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci USA 100: 8389–8394.
Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V et al (2015). Therapeutic effects of glatiramer acetate and grafted CD115(+) monocytes in a mouse model of Alzheimer's disease. Brain 138 (Pt 8): 2399–2422.
Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T et al (2013). IFN-gamma-dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain 136 (Pt 11): 3427–3440.
Lee BK, Magnusson C, Gardner RM, Blomstrom A, Newschaffer CJ, Burstyn I et al (2015). Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav Immun 44: 100–105.
Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM (2007). Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 130: 166–176.
Li Y, Du XF, Liu CS, Wen ZL, Du JL (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 23: 1189–1202.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523: 337–341.
Lueg G, Gross CC, Lohmann H, Johnen A, Kemmling A, Deppe M et al (2015). Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer's disease. Neurobiol Aging 36: 81–89.
Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH et al (2009). Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab 29: 921–932.
Marques F, Sousa JC, Correia-Neves M, Oliveira P, Sousa N, Palha JA (2007). The choroid plexus response to peripheral inflammatory stimulus. Neuroscience 144: 424–430.
Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H et al (2016). The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci USA 113: E1316–E1325.
Mesquita SD, Ferreira AC, Gao F, Coppola G, Geschwind DH, Sousa JC et al (2015). The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer's disease. Brain Behav Immun 49: 280–292.
Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M et al (2007). Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 10: 1544–1553.
Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K (2013). Causes, consequences, and reversal of immune system aging. J Clin Invest 123: 958–965.
Nimmerjahn A, Kirchhoff F, Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318.
Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M et al (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's-like pathology. Brain 139 (Pt 3): 891–907.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458.
Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ et al (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155: 1596–1609.
Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 109: E197–E205.
Petito CK, Adkins B (2005). Choroid plexus selectively accumulates T-lymphocytes in normal controls and after peripheral immune activation. J Neuroimmunol 162: 19–27.
Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S et al (2015). TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest 125: 2161–2170.
Radjavi A, Smirnov I, Derecki N, Kipnis J (2014). Dynamics of the meningeal CD4(+) T-cell repertoire are defined by the cervical lymph nodes and facilitate cognitive task performance in mice. Mol Psychiatry 19: 531–533.
Ragozzino D, Di Angelantonio S, Trettel F, Bertollini C, Maggi L, Gross C et al (2006). Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J Neurosci 26: 10488–10498.
Raposo C, Graubardt N, Cohen M, Eitan C, London A, Berkutzki T et al (2014). CNS repair requires both effector and regulatory T cells with distinct temporal and spatial profiles. J Neurosci 34: 10141–10155.
Raynor J, Karns R, Almanan M, Li KP, Divanovic S, Chougnet CA et al (2015). IL-6 and ICOS antagonize Bim and promote regulatory T cell accrual with age. J Immunol 195: 944–952.
Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S et al (2009). C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 10: 514–523.
Ron-Harel N, Segev Y, Lewitus GM, Cardon M, Ziv Y, Netanely D et al (2008). Age-dependent spatial memory loss can be partially restored by immune activation. Rejuvenation Res 11: 903–913.
Rook GA, Lowry CA, Raison CL (2011). Lymphocytes in neuroprotection, cognition and emotion: is intolerance really the answer? Brain Behav Immun 25: 591–601.
Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014). Transcranial amelioration of inflammation and cell death after brain injury. Nature 505: 223–228.
Sayed BA, Christy AL, Walker ME, Brown MA (2010). Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: a role for neutrophil recruitment? J Immunol 184: 6891–6900.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R et al (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: 691–705.
Schlager C, Korner H, Krueger M, Vidoli S, Haberl M, Mielke D et al (2016). Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 530: 349–353.
Sheng J, Ruedl C, Karjalainen K (2015). Most Tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43: 382–393.
Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE et al (2015). Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 35: 13029–13042.
Shrestha B, Paul D, Pachter JS (2014). Alterations in tight junction protein and IgG permeability accompany leukocyte extravasation across the choroid plexus during neuroinflammation. J Neuropathol Exp Neurol 73: 1047–1061.
Sipe GO, Lowery RL, Tremblay ME, Kelly EA, Lamantia CE, Majewska AK (2016). Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun 7: 10905.
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH (2007). Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 27: 10695–10702.
Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M et al (2016). Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 139 (Pt 4): 1265–1281.
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L et al (2013). A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33: 13460–13474.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N et al (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178.
Tremblay ME, Lowery RL, Majewska AK (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8: e1000527.
van der Geest KS, Abdulahad WH, Tete SM, Lorencetti P, Horst G, Bos NA et al (2014). Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp Gerontol 60: 190–196.
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29: 3974–3980.
Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F et al (2016). Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J Neurosci 36: 2827–2842.
Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A et al (2009). CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol 182: 3979–3984.
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R et al (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211: 1533–1549.
Yirmiya R, Goshen I (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25: 181–213.
Young KG, Maclean S, Dudani R, Krishnan L, Sad S (2011). CD8+ T cells primed in the periphery provide time-bound immune-surveillance to the central nervous system. J Immunol 187: 1192–1200.
Zhang J, Malik A, Choi HB, Ko RW, Dissing-Olesen L, MacVicar BA (2014). Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 82: 195–207.
Zhang X, Wu C, Song J, Gotte M, Sorokin L (2013). Syndecan-1, a cell surface proteoglycan, negatively regulates initial leukocyte recruitment to the brain across the choroid plexus in murine experimental autoimmune encephalomyelitis. J Immunol 191: 4551–4561.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N et al (2006). Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9: 268–275.
Acknowledgements
We thank Shirley Smith for editing the manuscript. We thank the members of the Center for Brain Immunology and Glia (BIG) for their insightful comments and critical evaluation of the results. Special thanks to Dr Antoine Louveau for contributing the cover image of meningeal lymphatic vessels for this issue. This work was primarily supported by a grant from the National Institute of Mental Health, NIH (MH096484) to JK.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Marin, I., Kipnis, J. Central Nervous System: (Immunological) Ivory Tower or Not?. Neuropsychopharmacol 42, 28–35 (2017). https://doi.org/10.1038/npp.2016.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.122
This article is cited by
-
A novel 4 immune-related genes as diagnostic markers and correlated with immune infiltrates in major depressive disorder
BMC Immunology (2022)
-
Identification of pathways that regulate circadian rhythms using a larval zebrafish small molecule screen
Scientific Reports (2019)
-
Inflammation Mediates Body Weight and Ageing Effects on Psychomotor Slowing
Scientific Reports (2019)
-
Networks of blood proteins in the neuroimmunology of schizophrenia
Translational Psychiatry (2018)
-
Cognitive impairment in rheumatoid arthritis: role of lymphocyte subsets, cytokines and neurotrophic factors
Clinical Rheumatology (2018)
