
Abstract
Recent research in neuroimmunology has revolutionized our understanding of the intricate interactions between the immune system and the central nervous system (CNS). The CNS, an “immune-privileged organ”, is now known to be intimately connected to the immune system through different cell types and cytokines. While type 2 immune responses have traditionally been associated with allergy and parasitic infections, emerging evidence suggests that these responses also play a crucial role in CNS homeostasis and disease pathogenesis. Type 2 immunity encompasses a delicate interplay among stroma, Th2 cells, innate lymphoid type 2 cells (ILC2s), mast cells, basophils, and the cytokines interleukin (IL)-4, IL-5, IL-13, IL-25, TSLP and IL-33. In this review, we discuss the beneficial and detrimental roles of type 2 immune cells and cytokines in CNS injury and homeostasis, cognition, and diseases such as tumors, Alzheimer’s disease and multiple sclerosis.
Similar content being viewed by others

Introduction
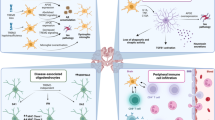
The mammalian central nervous system (CNS) has only a limited capacity for self-repair and renewal. Inflammation within the CNS can be detrimental to neurons. Therefore, the presence of immune cells in the CNS was long considered a hallmark of pathology. Furthermore, the absence of lymphatic vessels inside the brain parenchyma [1], the low to no expression of MHC II by brain-resident microglial cells during homeostasis, the absence of other antigen-presenting cells, and the presence of a blood−brain barrier that prevents immune-cell recruitment inside the brain led to and supported the notion that the brain is an “immunologically privileged” organ [2, 3]. Recent advances in the understanding of neuroimmune interactions in the brain and its borders have shifted our understanding of the role of the peripheral immune system in CNS homeostasis and disease [4, 5] (Fig. 1).

Neuroimmune circuitry in the brain and borders. Cytokines secreted by mast cells, ILC2s, and Th2s located in the meninges and choroid plexus modulate behavior and learning through receptors expressed by neurons and glia cells. Specifically, IL-4 can mediate its effect directly through the IL-4Rα expressed on GABAergic neurons, and astrocytes, in response to IL-4, produce brain-derived neurotropic factor (BDNF), a key molecule in learning and memory. Th2 cells accumulate in the choroid plexus, resulting in a local excess of IL-4 that acts on choroid plexus epithelial cells to produce CCL11, which has been correlated with the cognitive decline observed during aging. In addition, IL-13-deficient mice exhibit cognitive impairment similar to that observed in IL-4 knockout (KO) mice. IL-13 was also shown to be a synaptic protein in mouse and human brains, localized in the presynaptic membrane, whereas IL-13Rα1 is localized on the postsynaptic membrane. The engagement of IL-13 with type 2 receptors results in the phosphorylation of the NMDAR and AMPAR subunits and increases synaptic activity
Meninges, the three-layer protective membranes surrounding the brain and spinal cord, were traditionally considered passive barriers, but it is now clear that they are actively involved in regulating immune responses in the CNS. Single-cell profiling of meninges has revealed that both the leptomeninges (arachnoid mater and pia mater) and the dura mater are immune-rich tissues harboring cells of diverse lymphoid and myeloid lineages. Cerebrospinal fluid (CSF) from the brain can directly access the cranial meninges, where CNS-derived antigens are continuously sampled by dural sinus-associated antigen-presenting cells [6]. Moreover, within the meningeal dural layer, a functional lymphatic system drains antigens and macromolecules from the CNS to be sampled by the draining cervical lymph nodes [7, 8]. These findings have led to the understanding that within the meninges, the CNS harbors compartmentalized immune niches containing a rich repertoire of immune cells [9,10,11,12] that affect CNS homeostasis and pathologies [13, 14]. Elegant studies of novel mouse models have revealed that inflammation, in the context of sterile injury, is beneficial to the damaged CNS [15, 16]. Recent advances in the anatomical, molecular and cellular composition of the brain borders are reviewed elsewhere [4].
We now know that inflammation and the immune system are involved in almost all types of human acute and chronic diseases, including mental and physical health problems [17], and that they play a major role in enforcing homeostasis, as well as in the functional and structural integrity of the tissues, including the brain [18]. Homeostasis and inflammation are fundamentally connected, and most pathological states have a homeostatic counterpart [19]. In the spectrum of inflammatory responses, physiological inflammation occurs in the absence of infection or injury and is aimed at restoring the system to a homeostatic state. When this response is insufficient, type 2 inflammation-associated signals such as cytokines, chemokines, bioactive amines, and immune cells play a role in acute and chronic inflammation [20].
Type 2 response-associated immune cells, including T helper 2 (Th2) cells, innate lymphoid type 2 cells (ILC2), eosinophils, mast cells, basophils and alternatively activated macrophages, are associated with either a protective or a pathogenic phenotype depending on the circumstances [21]. Originally, it was thought that the primary role of type 2 immunity was to limit the consequences of type 1-driven inflammation. While it is true that type 1 inflammation caused by structural loss is often followed by a type 2 inflammatory response, it is also often initiated owing to a loss of function [20] caused by noxious substances such as allergens [22], xenobiotics, and parasitic worms [23] that can disrupt epithelial barriers [24]. Inflammation caused by structural loss (type 1), functional loss (type 2), and regulatory loss (type 3) can antagonize other types of responses. In some cases, the induction of type 2 immunity limits the inflammation caused by a Th1/Th17 response and is beneficial for the host. In the case of experimental autoimmune encephalomyelitis (EAE), interleukin-4 (IL-4)-induced type 2 immunity suppresses type 1 driven inflammation, thereby ameliorating EAE [25]. On the other hand, it is crucial for the host to mount appropriate immune responses against invading pathogens. For example, Leishmania major requires a Th1 response to clear the infection; therefore, in BALB/c mice, where CD4+ T cells differentiate into Th2 cells in response to injury, these cells are unable to control the infection, a result that is detrimental to the host. Moreover, pathogenic activation of type 2 immunity in response to harmful environmental stimuli can contribute to the development of diseases/conditions that include asthma, allergic rhinitis, atopic dermatitis, and allergies to drugs and foods [26], while excessive inhibition of cytotoxic type 1 immunity by IL-4- and IL-13-activated macrophages can promote tumor development as well as pathological fibrosis or organ scarring [27]. It is therefore crucial for the CNS to tightly regulate the induction of inflammation or to limit its pathogenic activation.
Cytokines involved in the initiation of the type 2 response
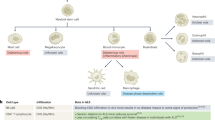
IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) are crucial cytokines that play a central role in the induction of type 2 inflammation. IL-33 belongs to the IL-1 family of cytokines and is released as an alarmin in response to cell injury or tissue damage. IL-33 acts on cells expressing the ST2 receptor, also known as interleukin 1 receptor-like 1 (IL1RL1) [28], including mast cells, ILC2s, Tregs, Th2 cells, eosinophils, basophils, dendritic cells (DCs), and macrophages (including CNS-resident microglia) [29]. IL-33 has a multifaceted role in homeostasis and pathology and is expressed by many cell types, including hematopoietic, stromal, and parenchymal cells in the CNS and its borders [6, 30].
IL-33 is highly expressed in developing brain astrocytes and oligodendrocytes [31, 32]. Astrocytes residing near redundant synapses release IL-33 to recruit microglia, promoting synapse engulfment by microglia. A disruption of the IL-33 axis leads to overactive brain circuitry, behavioral abnormalities and seizures owing to increased numbers of excitatory synapses [33], presumably because of microglial dystrophy. Moreover, IL-33 is upregulated in the mature brain by astrocytes upon Toll-like receptor (TLR) stimulation [34] and is constitutively expressed by oligodendrocytes [35]) and neurons [36]. The IL-33/ST2 axis has been implicated in many CNS-related diseases and conditions, including Alzheimer’s disease [37, 38], age-related macular degeneration [39], multiple sclerosis (MS) [40] and EAE [41,42,43], stroke [44], CNS injury [35, 45, 46], and pain [47].
Findings from studies in mice suggest that IL-33 is protective in models of stroke [44] and CNS injury [35]. In the healthy brain, IL-33 is highly expressed by oligodendrocytes. Upon injury, IL-33 is released through an unknown mechanism and promotes monocyte recruitment to the injured site [35]. Mice lacking IL-33 have impaired recovery following spinal cord injury (SCI). Upon its release, IL-33 can also activate ILC2s residing in the meninges, which in turn upregulate IL-5 and IL-13, inducing type 2 immunity [46].
MS patients have elevated IL-33 levels in the brain and plasma [40]. This increase in IL-33 might be a compensatory mechanism toward Th1/Th17-mediated inflammation. ST2 expression increases in the spinal cord after EAE, and ST2-deficient mice have exacerbated EAE [43]. IL-33 treatment can reduce IL-17 and IFN-y levels, most likely by inducing IL-5 and IL-13 [43]. Another possible role of the IL-33/ST2 axis in EAE is mediated through CNS regulatory T cells (Tregs), which also express ST2. The deletion of ST2 in Foxp3+ Tregs leads to EAE exacerbation by diminishing the ability of Tregs to suppress IL-17A-secreting gamma-delta T cells [48]. IL-33 function can be modulated via cleavage by proteases and tryptases secreted by neutrophils and by activated mast cells. Cleaved IL-33 is a potent activator of ILC2s and eosinophils [49]. Importantly, mast-cell-derived IL-33 activates ILC2s in the meninges, which in turn promote a Th2 response in the SJL mouse model of MS, limiting the progression of the disease [42].
IL-33 function in chronic neurodegenerative diseases is less well understood. The first evidence comes from human patients. IL-33 is decreased in the brains of patients with Alzheimer’s disease. Moreover, IL-33 polymorphisms modulate the risk of Alzheimer’s disease, with some rare single-nucleotide polymorphisms (SNPs) found to be protective [37]. Soluble ST2, a decoy receptor for IL-33, is upregulated in the serum of patients with mild cognitive impairment. Intraperitoneal administration of IL-33 enhances microglial phagocytosis and improves synaptic impairment and Aβ aggregation in APP/PS1 mice [38].
In conclusion, IL-33 plays a crucial role in inflammation and homeostasis, and its multifaceted role is still being researched. The involvement of IL-33 in CNS-related diseases, including Alzheimer’s disease, MS and EAE, highlights its potential as a therapeutic target. Further work is needed to better understand the mechanisms underlying the role of IL-33 and to identify its potential as a therapeutic agent.
The functions of other type 2 immunity inducers in the CNS are less well characterized. IL-25, for example, may be involved in EAE and MS: its expression in microglia is increased after EAE induction, and it is important in maintaining the blood−brain barrier and limiting Th17-mediated inflammation. Accordingly, IL-25-deficient mice are highly susceptible to EAE [50]. The protective role of IL-25 is also mediated through the upregulation of IL-13 by Th2 cells and potentially by ILC2s, which inhibit IL-23, IL-1b, and IL-6 expression in DCs. Furthermore, human brain capillaries express IL-25, which is downregulated in tissues near severe MS lesions [51].
Th2 cells and effector cytokines
IL-4 and IL-13 were among the first cytokines identified, and their functions often overlap. IL-4 can elicit its function by binding with two types of receptors: the type 1 receptor, which is expressed mainly on hematopoietic cells and uses IL-4Rα and the common gamma chain, and the type 2 receptor, which is expressed on nonhematopoietic cells and possesses two subunits, IL-4Rα and IL-13Rα1. The cytokine IL-13, on the other hand, can bind only with the type 2 receptor. The binding of IL-4 with the type 1 complex phosphorylates JAK1/3, which in turn phosphorylates tyrosines within IL-4Rα cytoplasmic domains, creating docking sites for STAT6 and insulin receptor substrate 2 (IRS2). In naïve CD4+ T cells, STAT6 induces the expression of GATA3 and thus leads to their differentiation into Th2 cells. In B cells, STAT6 induces class switching to IgE and promotes alternative activation of macrophages [52]. The type 2 receptor is associated with JAK1 and TYK2, leading to STAT6 phosphorylation, homodimerization, and nuclear translocation [53]. IL-13, in addition to binding with the type 2 receptor, binds with the decoy receptor IL-13Rα2, which lacks a cytoplasmic domain and therefore has a higher affinity for IL-13. IL-13Rα2 is often overexpressed in malignant tumors, especially in glioblastoma multiforme (GBM). Newer findings suggest that in addition to IL-13 signaling, IL-13Rα2 can attenuate IL-4 signaling by inhibiting the dimerization of IL-4Rα with second subunits. Furthermore, IL-13Rα2 promotes transforming growth factor beta (TGF-b) production in monocytes and thus promotes tissue fibrosis [54]. IL-4 and IL-13 are important in reducing IL-1-induced inflammation by increasing the expression of IL-1R2, a decoy receptor for IL-1β that limits inflammation [55].
Recent research has revealed roles of IL-4 and IL-13 in the CNS beyond their established functions in immune regulation and inflammation. T cells have long been implicated in the modulation of learning and behavior [16, 56]. In the healthy adult CNS, T cells reside mainly in the meninges and are not found in the brain parenchyma [6]. IL-4 and IL-13 produced by meningeal T cells are involved in learning behaviors [57, 58]. IL-4-deficient mice were shown to exhibit learning deficits [58]. Similarly, SCID and CD4-depleted mice were found to exhibit impaired learning in different behavioral tasks. IL-4Rα is expressed in some brain-resident cells, including neurons. Earlier studies suggested that IL-4 mediates its effect on learning through astrocytic IL-4Rα [58,59,60], whereby astrocytes, in response to IL-4, produce brain-derived neurotropic factor (BDNF), a key molecule in learning and memory. A new line of evidence suggests that cytokines can directly modulate neuronal responses through receptors expressed on neurons [61,62,63]. Similarly, a recent study showed that IL-4 can mediate its effect directly through IL-4Rα expressed on GABAergic neurons. Conditional ablation of IL-4Rα on GABAergic neurons but not microglia or macrophages resulted in impaired memory. Moreover, memory deficits were restored after the injection of wild-type CD4 cells into SCID mice but not if transferred T cells were IL-4 deficient [64].
Resident T cells in the choroid plexus are another source of IL-4 in the brain [65]. As mice age, Th2 cells accumulate in the choroid plexus, resulting in a local excess of IL-4 that acts on choroid plexus epithelial cells to produce CCL11 [65]. This increase in CCL11 has been correlated with the cognitive decline observed during aging [66] and has also been implicated in the cognitive syndrome observed in patients with long COVID-19 [67].
In addition to IL-4, other Th2-derived cytokines have also been implicated in the modulation of behavior and learning. IL-5 was shown to enhance cognitive function, specifically spatial recognition, in mouse models of Alzheimer’s disease [68].
IL-13-deficient mice exhibit cognitive impairment similar to that observed in IL-4 knockout (KO) mice [57]. While potential sources of IL-4 in the CNS include mostly immune cells, such as Th2 cells, mast cells and basophils, IL-13 was also shown to be a synaptic protein in mouse and human brains. This cytokine is localized in the presynaptic membrane, whereas IL-13Rα1 is localized on the postsynaptic membrane. The engagement of IL-13 with type 2 receptors results in the phosphorylation of the NMDAR and AMPAR subunits and increases synaptic activity [69].
In addition to modulating mammalian behavior, type 2 cytokines have been implicated in CNS diseases and conditions, including depression [70], injury [15], Alzheimer’s disease [71], MS, stroke [72], and traumatic brain injury [69, 73]. IL-13 expression is enhanced during traumatic brain injury in human neurons, probably as a compensatory protective mechanism in the case of excitatory death [69]. The neuroprotective role of CNS-infiltrating Th2 cells during SCI or optic nerve crush is independent of the binding of the T-cell receptor (TCR) with its cognate antigens [15]. Th2 cells promote functional recovery after spinal cord and optic nerve injury [15]. The protective effect of IL-4 has also been demonstrated in a mouse model of Alzheimer’s disease, where the injection of IL-4 alone or of IL-4 + IL-13 induced the proliferation of Arg1+ microglia and the clearance of plaques in APP/PS1 mice [71, 74]. Other studies, however, have found contraindicatory results [75].
The protective role of type 2 immunity in Th1/Th17-mediated diseases is supported by studies using knockout mouse models. For example, mice lacking IL-10 are more susceptible to the development of severe EAE than wild-type mice, indicating that IL-10 plays a critical role in regulating the immune response in this disease. In contrast, IL-4 KO mice showed no such effect [76, 77]. This may be due to the different polarizing effects of IL-4 and IL-10 [78]. IL-4 induces macrophage polarization through STAT6, whereas IL-10 acts through STAT3 signaling. Interestingly, IL-10-activated microglia closely resemble human microglia in Nasu–Hakola disease, which is caused by mutations in the TREM2 or DAP12 genes [79].
Mast cells
Mast cells have long been considered master regulators of allergic inflammation. We are now also beginning to recognize their role in homeostasis [80] and in responses such as the recruitment of neutrophils during skin inflammation [81], host defense against bacteria [82], inflammatory pain [83], and immunosuppression [84]. Mast cells in the healthy CNS reside in the meninges, mostly in the dura, and presumably also in the brain parenchyma in small numbers [85,86,87]. Originating from late erythro-myeloid progenitors [88], mast cells mature in tissues and acquire unique gene signatures in different niches [89]. In addition to their expression of the canonical mast-cell tryptase and proteases, mast cells express high levels of IL-4 and IL-13. They also exhibit immunoregulatory functions, and after IL-33 stimulation, they can suppress allergic inflammation by secreting IL-2, which in turn promotes Treg expansion [90], thus making them prime candidates for mounting type 2 responses [91].
In the brain, mast cells were first identified in 1890, near MS brain lesions [92]. Their involvement in the pathogenesis of MS is further supported by human studies. Mast cell-specific tryptase is elevated in the CSF of MS patients [93]. Their role in the pathogenesis of MS, however, is still not fully understood [94]. In histochemical studies, brain mast cells seemed to lack expression of c-Kit and FCER1A, canonical markers of tissue mast cells [95, 96], making the study of brain-resident mast-cell function particularly challenging.
Early studies on mast-cell involvement in the pathogenesis of EAE development, carried out largely on W/Wv and KitW-sh/W-sh mouse models, showed that mast-cell-deficient W/Wv mice demonstrated delayed onset and reduced severity of EAE [97]. The transplantation of bone marrow-derived mast cells restored the clinical course of this disease. Notably, transplantation failed to reconstitute brain mast cells, suggesting that peripheral mast cells might play a role in such restoration [98]. In contrast, Kitw-sh mice demonstrated exacerbated EAE disease, and peripheral restoration of mast cells via bone marrow-derived mast-cell transplantation was insufficient to change its clinical course, indicating a complex role for mast cells in EAE [98]. Extensive studies have aimed to elucidate the role of meningeal mast cells in the efficient infiltration of inflammatory T cells into the CNS during EAE development. Mast cells in the meninges are strategically located near the meningeal vasculature and dural sinuses. They promote CNS recruitment of immune cells by secreting proinflammatory mediators such as tumor necrosis factor, which can activate nearby endothelial cells and enhance leukocyte adhesion and extravasation into the CNS [99]. Meningeal mast cells in EAE mice have been shown to play a crucial role in the efficient CNS infiltration of inflammatory T cells. The transfer of mast cells into W/Wv mice was shown to reconstitute meningeal mast cells, which alleviated EAE severity and delayed EAE onset, thus highlighting the importance of these cells in EAE pathogenesis. Meningeal mast cells also recruit neutrophils to the site of inflammation, which can further exacerbate tissue damage in the CNS [99].
Mast cells have been found to play critical roles in CNS homeostasis, including the modulation of mammalian behavior. Given their identification as immune cells containing heparin and histamine, mammalian mast cells are morphologically consistent with the mast cells observed in ancient organisms such as tunicates and crustaceans, suggesting that these cells coevolved with the nervous system. Mast cell function is tightly regulated by neurotransmitters and neuropeptides such as acetylcholine, gamma-aminobutyric acid (GABA), glutamate, dopamine, substance P, vasoactive intestinal peptide (VIP), and calcitonin gene-related peptide (CGRP) [100]. CGRP-mediated mast-cell activation is particularly important in CNS homeostasis; for example, neurogenic inflammation leading to migraine is mediated through CGRP. Moreover, chemical degranulation of meningeal mast cells with Compound 48/80 leads to prolonged excitation of nociceptors in the meninges, with downstream trigeminal activation [101]. This neuroimmune communication seems to be bidirectional. In Kitw-sh mice, mast-cell deficiency has been associated with behavioral abnormalities and an anxiety-like phenotype. In addition, pharmacological inhibition of mast-cell degranulation by intracerebroventricular injection of disodium cromoglycate increased anxiety-like behavior in mice. No significant effect was observed after intraperitoneal injection, suggesting the importance of mast cells residing in the CNS [102].
New evidence suggests that mast cells may actually be protective under certain conditions. Studies in Kitw-sh mast-cell-deficient mice have shown that mast cells may be protective in models of SCI and mechanical brain injury [103, 104]. Compared to wild-type controls, Kitw-sh mice exhibited significantly higher T-cell infiltration and reduced functional recovery in these experiments. It was suggested that these effects are mediated through MCP4 cleavage of MCP-1, IL-6 and IL-13, and mice lacking mMCP4 showed defects in functional recovery after SCI [104].
Despite the findings discussed above, there is a need for critical reevaluation, as the lack of mast-cell-specific mouse models has contributed to ambiguity in the field. The first-generation mast-cell-deficient mouse model, W/Wv, has severe hematopoietic perturbations, including anemia, as well as reduced γδ T cells, basophils, and neutropenia. Similarly, KitW-sh/W-sh mice demonstrate enhanced myelopoiesis and a subsequent increase in neutrophils and basophils. Studies with new mast-cell-deficient models that are not based on those first-generation models have failed to replicate many findings attributed to mast cells, including their role in EAE [105]. In the CPA3 -/- mouse model, which lacks connective tissue mast cells but also has reduced numbers of basophils in the blood, researchers found no involvement of mast cells in autoimmune diseases and failed to reproduce differences in EAE susceptibility in W/Wv mice [106].
ILC2s
Recent findings on ILC2s have shed light on their diverse functions in various biological processes. ILC2s are highly enriched in barrier surfaces, where they play crucial roles in initiating type 2 immune responses in adipose tissue homeostasis [107], lung homeostasis and inflammation [108, 109], and gut barrier functions [110].
The majority of tissue-resident ILC2s in adults are generated de novo during the postnatal period. Owing to their transcriptional and functional similarities, ILC2s were initially considered to be an innate counterpart of Th2 cells. However, other subtypes of ILC2s have since been described, such as the IL-10-producing killer cell lectin-like receptor G1 (KLRG1)+ ILC2s, which suppress Th2 responses and induce tolerance [111], and type 2 induced inflammatory ILC2s (iILC2s), which are recruited to the lungs from the intestines in response to exposure to IL-25 or to parasitic worms [112]. ILC2s are activated by IL-33, IL-25, and TSLP in the lungs, gut, and skin [113,114,115]. Moreover, skin ILC2s express IL-4Ra and can be activated by basophil-derived IL-4 in inflamed mouse and human skin [116] as well as in the inflamed lung, where activated ILC2s promote eosinophil infiltration through the secretion of IL-5 and IL-13 [117].
In addition to their roles in type 2 immune responses and tissue homeostasis, ILC2s play a role in hematopoiesis. ILC2s are present in bone marrow, including skull bone marrow, and can stimulate hematopoiesis. In the context of 5-fluorouracil (5-FU)-induced stress, B-cell progenitor-derived IL-33 activates myeloid differentiation primary response 88 (MyD88)-mediated secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) in ILC2s to support myeloid hematopoiesis. Moreover, ILC2s can expand and become activated during stress, upregulating programmed cell death protein (PD)-1, ST2 and cluster of differentiation 25 (CD25) and downregulating IL-7Rα. In granulocyte-macrophage colony-stimulating factor (GM-CSF) KO mice, the transplantation of wild-type ILC2s was found to be sufficient to restore hematopoiesis [118]. IL-33 has also been shown in certain contexts to induce IL-10 expression and downregulate type 2 cytokine production in lung ILC2s [111].
ILC2 function is tightly regulated by the nervous system through the action of neurotransmitters. Both peptidergic and nonpeptidergic neurotransmitters, such as neuromedin U (NMU) and VIP, promote ILC2 function [119, 120]. Conversely, the engagement of B2 adrenergic receptors with epinephrine inhibits ILC2 proliferation and function [121]. During inflammation, this neuroimmune circuit becomes even more complicated, as other cells can affect receptor expression on ILC2s and further modulate their activation and function, such as basophils, which have been shown to affect ILC2 function through the regulation of neuromedin B expression on ILC2s during lung infection with the nematode Nippostrongylus brasiliensis [122].
The interplay between ILC2s and the nervous system is further exemplified by the role of CGRP in regulating ILC2 function. CGRP and its receptors are expressed by ILC2s in barrier tissues such as meninges and the lungs, where excess CGRP can dampen IL-33-induced inflammation and inhibit the type 2 cytokine production and proliferation of ILC2s during airway inflammation and parasitic infection [109, 123]. Similarly, in the context of meningeal bacterial inflammation, CGRP released from nociceptors dampens macrophage activation and recruitment of neutrophils and monocytes [124]. Interestingly, nociceptor ablation in the meninges results in increased neutrophil and monocyte recruitment and protects the brain from invading pathogens. Furthermore, in the context of type 2 immunity, IL-5 produced by activated ILC2s and Th2 cells can activate nociceptors through IL-5R and induce the release of VIP, creating an inflammatory circuit loop that promotes allergic inflammation.
ILC2s were also shown to be present in the spinal dura and choroid plexus, albeit to a lesser extent than in the cranial meninges. Interestingly, no ILC2s were found in the brain parenchyma or perivascular space [46]. In the dura, ILC2s are mainly located near the dural sinuses, where they are in close proximity to IL-33-expressing fibroblast-like cells, similar to the situation observed in lung and white adipose tissue [125]. During lung infection, ILC2- and Th2-derived IL-13 promotes the expansion of these stromal cells, which in turn upregulate IL-33, creating a positive feedback loop [125].
Meningeal ILC2s express Thy1.2, ST2, CD25, spinocerebellar ataxia type 1 (Sca10), and low levels of c-Kit and IL-7Rα and are major producers of IL-13 in tissues. As tissue-resident cells, ILC2s exhibit tissue-specific receptor expression patterns, which are imprinted by local signals [126]. Likewise, dural ILC2s are distinct from lung ILC2s. The basal activation state of meningeal ILC2s is lower than that of lung-resident cells [46]. The proximity to brain-derived signals may explain the differences in basal activation observed between the two tissue types. ILC2 precursors originate from bone marrow. Future studies should determine the functional and phenotypic differences between blood-derived ILC2s and the adjacent bone marrow.
The role of ILC2s in neuroinflammation is an emerging field of study. ILC2 function is best characterized in the spinal cord injury model. After SCI, cranial meningeal ILC2s become activated in an IL-33-dependent manner, produce IL-5 and IL-13, and upregulate CGRP and its receptors. This effect is abolished in IL-33 KO mice. The reconstitution of IL-33 KO mice with wild-type ILC2s restores the protective effect of meningeal ILC2s, leading to better functional recovery and reduced lesion area [46]. Interestingly, ILC2s also accumulate in the injured spinal cord 10 days after injury, but the origins of these cells and their relative contributions from local meningeal bone marrow and circulating progenitors are unknown.
ILC2 functions are also linked to cognition. In a mouse model of Alzheimer’s disease (3xTg-AD mice), defects in meningeal ILC2s, including a reduction in cell number and functional deficits in IL-5 production, were identified [68]. During aging, ILC2s accumulate in the meninges and choroid plexus. ILC2s are practically absent in the choroid plexus of young mice. Aged choroid plexus ILC2s are quiescent at steady state, but upon stimulation, they produce IL-5 and IL-13. The transfer of activated choroid plexus ILC2s was shown to increase the cognitive performance of mice after 1 week [127].
A correlation has also been observed between EAE susceptibility and ILC2 numbers in the brain and meninges, with EAE-resistant mice having larger numbers [128]. Also found to have decreased numbers of ILC2s are W/Wv mice, which lack mast cells owing to the disruption of c-kit signaling. The relationship between ILC2s and mast cells in EAE pathogenesis requires further investigation.
Tumors
Glioblastoma multiforme (GBM) is a highly aggressive brain tumor that can evade the surveillance mechanisms of the immune system, making it difficult to treat. The type 2 immune response is a complex mechanism that can be either beneficial or detrimental to cancer growth, depending on the context [129, 130]. On the one hand, the response can promote tumor dissemination and metastasis through the action of CD4+ Th2 effector cells, which regulate the pro-tumor properties of tumor-associated macrophages via IL-4 expression [131, 132], and IL-4 is upregulated in many mouse and human tumors [133]. Although their role in pathogenesis is still unknown, their enrichment in human and mouse GBM suggests that these effector cells may play a role in tumor growth [134]. Type 2 immunity is often regarded as an unfavorable antitumor response since it allows cancer cells to evade the immune system.
However, recent human epidemiological data suggest that the type 2 response can be an effective defense mechanism against cancer. For instance, there is an inverse correlation between allergy and the occurrence of glioblastoma (GBM) [135]. In addition, asthma-associated SNPs in IL-4R, IL-13, and STAT6 gene loci are inversely correlated with the occurrence of GBM [136, 137]. Eliciting a type 2 response in the early stages of different cancers can be protective [138, 139]. This effect can be mediated through various mechanisms, including the recruitment of eosinophils in an IL-5-dependent manner [140, 141], TSLP-mediated Th2 polarization [138], and IL-4-mediated reorganization of tumor vasculature with subsequent hypoxia and cancer cell death [142].
Another newly recognized type 2 antitumoral effect is mediated through IgE. IgE is an ancient immunoglobulin thought to have evolved to protect against infections caused by large parasites, such as helminths. Although IgE increases in abundance in the presence of helminth infections, it is not critical for protective immunity against these parasites, as it has been found to respond to environmental toxins and xenobiotics that may be carcinogens [143]. Early establishment of an IgE response via IL-4 toward carcinogenic DNA-damaging environmental xenobiotic 9,10-dimethylbenz-A-anthracene (DMBA) restricts tumor growth in a basophile FC epsilon receptor (FcεRIα)-dependent manner [144]. Furthermore, FcεRIα is associated with positive survival in the majority of human tumors, including GBM [145], and treatment with omalizumab, a monoclonal antibody against IgE, might be associated with a higher risk of developing cancer [146]. In addition, IL-13Rα2, a restrictive receptor for IL-13, is overexpressed in the majority of GBM patients, making it a prime candidate for chimeric antigen receptor (CAR) T-cell therapy [147]. These data indicate that GBM actively suppresses type 2 responses to promote tumor growth.
The manipulation of the meningeal lymphatic system has shown promise in enhancing immune responses against GBM. For example, ectopic expression of vascular endothelial growth factor (VEGF)-C enhances the meningeal lymphatic network and the drainage of tumor antigens into the deep cervical lymph nodes. The treatment of mice with VEGF-C was shown to result in the clearance of GBM cells, and this effect was dependent on both CD4 and CD8 T cells, as the depletion of these T cells abolished VEGF-C-driven protection [148].
GBM can further subvert immune activation by recruiting myeloid suppressor cells through the production of the chemoattractant CXCL12 [149]. The CXCL12 receptor CXCR4 is widely expressed on myeloid cells and is essential for their retention in bone marrow and for recruitment into the tissue. The disruption of the local CXCL12–CXCR4 axis in the bone marrow results in the rapid recruitment of monocytes and neutrophils into the meninges [150]. Local bone marrow in the skull and in vertebral columns are directly connected to the meninges via vascularized bone channels [151,152,153]. These adjacent bone marrow sites act as myeloid and lymphoid cell reservoirs in homeostasis, aging, and acute and chronic CNS pathologies such as stroke and EAE [150, 151, 154]. During stroke, skull bone marrow neutrophils are the primary responders in the initial stages of damage [151]. Moreover, in EAE, local bone marrow-derived monocytes have been shown, based on their gene signature, to be less inflammatory, raising the possibility of discrete functions of adjacent bone marrow and blood-derived cells [150]. Moreover, recent findings suggest that this route is bidirectional and that the brain can directly regulate the hematopoietic niche in adjacent bone marrow sites in mice as well in humans [155,156,157]. CSF from the brain can directly access adjacent bone marrow via these channels, an axis shown to be important in EAE and meningitis. Similarly, GBM-derived CXCL12 can aid in the recruitment of adjacent bone marrow-derived myeloid suppressor cells through those channels. These findings shed light on the complex interplay among the meningeal lymphatic system, the hematopoietic niche and GBM pathogenesis, suggesting new avenues for GBM immunotherapy.
Conclusions and perspectives
Type 2 immunity is an adaptive immune response commonly associated with allergic inflammation or helminth parasite infection. It plays a crucial role in tissue repair and involves complex coordination between cells and cytokines. The induction of type 2 immunity in response to a loss of function aims to eliminate perturbators by inducing peristalsis, diarrhea, vomiting, itching, and sneezing and by complementing a basal function. Type 2 immunity also plays roles in glucose homeostasis [158], thermogenesis [159], and insulin resistance [160]. Mounting evidence suggests, moreover, that type 2 immune responses in the brain and brain borders could shape adaptive and dysfunctional neurological processes. Type 2 cytokines are crucial for maintaining CNS homeostasis. Two cytokines involved in the initiation of type 2 responses, IL-33 and IL-25, are highly enriched in the CNS. Moreover, effector cytokines such as IL-4 and IL-13 can be secreted by different cell types in the healthy CNS and act as neuromodulators by binding with cognate receptors expressed by CNS-resident cells, including neurons. The type 2 response also often limits the detrimental consequences of type 1/type 3 inflammation and aims to restore tissue homeostasis. Emerging evidence suggests that Th2 cells are beneficial for the damaged CNS. Dampening the type 2 response by knocking out cytokines, receptors or cells involved in the response often leads to an exacerbation of inflammation and pathology.
While in this review we have focused on the type 2 responses initiated at the brain borders or in the CNS parenchyma itself, growing evidence suggests that the peripheral tissue−CNS axis is implicated in many CNS disorders. In the case of the gut-brain axis, for example, different gut microbiota affect mammalian behavior and even the progression of neurodegenerative and autoimmune diseases [161]. Similarly, systemic inflammation induced by lipopolysaccharide (LPS) or IL-1b can modulate behavior [162]. However, less is known about how systemic type 2 pathological activation modulates CNS homeostasis and disease progression. People with asthma and other allergic conditions are less likely to develop GBM [137]. Some reports suggest a correlation between atopy and neurobehavioral conditions [163,164,165,166] and headaches [167, 168]. Moreover, there is a critical need to re-evaluate the conflicting data regarding the functions of immune cells involved in the type 2 response using better tools. Future studies should determine the extent to which type 2 immunity contributes to these effects and identify potential therapeutic targets.
References
Sandrone S, Moreno-Zambrano D, Kipnis J, van Gijn J. A (delayed) history of the brain lymphatic system. Nat Med. 2019;25:538–40.
Louveau A, Harris TH, Kipnis J. Revisiting the concept of CNS immune privilege. Trends Immunol. 2015;36:569–77.
Nicholas MK, Stefansson K, Antel JP, Arnason BGW. An in vivo and in vitro analysis of systemic immune function in mice with histologic evidence of neural transplant rejection. J Neurosci Res. 1987;18:245–57.
Rustenhoven J, Kipnis J. Brain borders at the central stage of neuroimmunology. Nature. 2022;612:417–29.
Alves de Lima K, Rustenhoven J, Kipnis J. Meningeal immunity and its function in maintenance of the central nervous system in health and disease. Annu Rev Immunol. 2020;38:597–620.
Rustenhoven J, Drieu A, Mamuladze T, de Lima KA, Dykstra T, Wall M, et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell. 2021;184:1000–1016.e27.
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21:1380–91.
Antila S, Karaman S, Nurmi H, Airavaara M, Voutilainen MH, Mathivet T, et al. Development and plasticity of meningeal lymphatic vessels. J Exp Med. 2017;214:3645–67.
Croese T, Castellani G, Schwartz M. Immune cell compartmentalization for brain surveillance and protection. Nat Immunol. 2021;22:1083–92.
Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci. 2017;20:1300–9.
Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. 2018;48:380–395.e6.
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22:1021–35.
Salvador AF, de Lima KA, Kipnis J. Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol. 2021;21:526–41.
Salvador AFM, Kipnis J. Immune response after central nervous system injury. Semin Immunol. 2022;59:101629.
Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. Am Soc Clin Investig. 2015;125:699–714. https://www.jci.org/articles/view/76210/figure/10.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–75.
Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25:1822–32.
Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35.
Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–27.
Medzhitov R. The spectrum of inflammatory responses. Science. 2021;374:1070–5.
Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15:271–82.
Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008;454:445–54.
Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. 2013;13:607–14.
Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21:739–51.
Shaw MK, Lorens JB, Dhawan A, DalCanto R, Tse HY, Tran AB, et al. Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J Exp Med. 1997;185:1711–4. https://pubmed.ncbi.nlm.nih.gov/9151908/.
Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature 2012;484:465–72.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T Helper type 2-associated cytokines. Immunity 2005;23:479–90.
Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol. Rev. 2018;281:154–68.
Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–89.
He D, Xu H, Zhang H, Tang R, Lan Y, Xing R, et al. Disruption of the IL-33-ST2-AKT signaling axis impairs neurodevelopment by inhibiting microglial metabolic adaptation and phagocytic function. Immunity. 2022;55:159–173.e9.
Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33–LacZ gene trap reporter strain. J Immunol. 2012;188:3488–95.
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269–73.
Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT. Induction of IL-33 expression and activity in central nervous system glia. J Leukoc Biol. 2008;84:631–43.
Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron. 2015;85:703–9.
Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell. 2020;182:388–403.e15.
Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease. Mol Psychiatry. 2009;14:1004–16.
Fu AKY, Hung KW, Yuen MYF, Zhou X, Mak DSY, Chan ICW, et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci USA. 2016;113:E2705–13.
Xi H, Katschke KJ Jr, Li Y, Truong T, Lee WP, Diehl L, et al. IL-33 amplifies an innate immune response in the degenerating retina. J Exp Med. 2016;213:189–207.
Christophi GP, Gruber RC, Panos M, Christophi RL, Jubelt B, Massa PT. Interleukin-33 upregulation in peripheral leukocytes and CNS of multiple sclerosis patients. Clin Immunol. 2012;142:308–19.
Li M, Li Y, Liu X, Gao X, Wang Y. IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol. 2012;247:25–31.
Russi AE, Ebel ME, Yang Y, Brown MA. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proc Natl Acad Sci USA. 2018;115:E1520–9.
Jiang HR, Milovanović M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur J Immunol. 2012;42:1804–14.
Korhonen P, Kanninen KM, Lehtonen Š, Lemarchant S, Puttonen KA, Oksanen M, et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun. 2015;49:322–36.
Pomeshchik Y, Kidin I, Korhonen P, Savchenko E, Jaronen M, Lehtonen S, et al. Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun. 2015;44:68–81.
Gadani SP, Smirnov I, Wiltbank AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med. 2016;214:285–96.
Verri WA, Guerrero ATG, Fukada SY, Valério DA, Cunha TM, Xu D, et al. IL-33 mediates antigen-induced cutaneous and articular hypernociception in mice. Proc Natl Acad Sci USA. 2008;105. https://doi.org/10.1073/pnas.0712116105.
Hemmers S, Schizas M, Rudensky AY. T reg cell–intrinsic requirements for ST2 signaling in health and neuroinflammation. J Exp Med. 2020;218:e20201234.
Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. 2014;111:15502–7.
Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–70.
Sonobe Y, Takeuchi H, Kataoka K, Li H, Jin S, Mimuro M, et al. Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase Cϵ-dependent manner. J Biol Chem. 2009;284:31834.
Shimoda K, van Deursent J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted State6 gene. Nature. 1996;380:630–3.
McCormick SM, Heller NM. Commentary: IL-4 AND IL-13 receptors and signaling. Cytokine. 2015;75:38.
Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat Med. 2006;12:99–106.
Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–5.
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: Implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA. 2004;101:8180–5.
Brombacher TM, Nono JK, De Gouveia KS, Makena N, Darby M, Womersley J, et al. IL-13-mediated regulation of learning and memory. J Immunol. 2017;198:2681–8.
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–80.
Brombacher TM, Berkiks I, Pillay S, Scibiorek M, Moses BO, Brombacher F. IL-4R alpha deficiency influences hippocampal-BDNF signaling pathway to impair reference memory. Sci Rep. 2020;10:16506.
Brombacher TM, Ajonijebu DC, Scibiorek M, Berkiks I, Moses BO, Mpotje T, et al. IL-4Rα deletion disrupts psychomotor performance and reference memory in mice while sparing behavioural phenotype associated with spatial learning. Brain Behav Immun. 2021;92:157–64.
Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol. 2020;21:1421–9.
Chen C, Itakura E, Nelson GM, Sheng M, Laurent P, Fenk LA, et al. IL-17 is a neuromodulator of Caenorhabditis elegans’ sensory responses. Nature. 2017;542:43–8.
Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature. 2016;535:425–9.
Herz J, Fu Z, Kim K, Dykstra T, Wall M, Li H, et al. GABAergic neuronal IL-4R mediates T cell effect on memory. Neuron. 2021;109:3609–3618.e9.
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci USA. 2013;110:2264–9.
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–4. https://pubmed.ncbi.nlm.nih.gov/21886162/.
Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee MH, Wood J, et al. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv. 2022. https://doi.org/10.1101/2022.01.07.475453v1.
Fung ITH, Zhang Y, Shin DS, Sankar P, Sun X, D’Souza SS, et al. Group 2 innate lymphoid cells are numerically and functionally deficient in the triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2021;18:152.
Li S, Olde Heuvel F, Rehman R, Aousji O, Froehlich A, Li Z, et al. Interleukin-13 and its receptor are synaptic proteins involved in plasticity and neuroprotection. Nat Commun. 2023;14:200.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP + PS1 bigenic mice. FASEB J. 2010;24:3093–102.
Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, et al. Peripheral administration of IL-13 induces anti-inflammatory microglial/macrophage responses and provides neuroprotection in ischemic stroke. Neurotherapeutics. 2019;16:1304–19.
Miao W, Zhao Y, Huang Y, Chen D, Luo C, Su W, et al. IL-13 ameliorates neuroinflammation and promotes functional recovery after traumatic brain injury. J Immunol. 2020;204:1486–98.
Kawahara K, Suenobu M, Yoshida A, Koga K, Hyodo A, Ohtsuka H, et al. Intracerebral microinjection of interleukin-4/interleukin-13 reduces β-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience. 2012;207:243–60.
Chakrabarty P, Tianbai L, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol Neurodegener. 2012;7:36.
Bettelli E, Prabhu Das M, Howard ED, Weiner HL, Sobel RA, Kuchroo VK. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice1. J Immunol. 1998;161:3299–306.
Falcone M, Rajan AJ, Bloom BR, Brosnan CF. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice1. J Immunol. 1998;160:4822–30.
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61.
Zhou Y, Tada M, Cai Z, Andhey PS, Swain A, Miller KR, et al. Human early-onset dementia caused by DAP12 deficiency reveals a unique signature of dysregulated microglia. Nat Immunol. 2023;24:545–57.
Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood. 2011;118:5383–93.
Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Köhler A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. 2011;34:973–84.
Starkl P, Watzenboeck ML, Popov LM, Zahalka S, Hladik A, Lakovits K, et al. IgE effector mechanisms, in concert with mast cells, contribute to acquired host defense against Staphylococcus aureus. Immunity. 2020;53:793–804.e9.
Green DP, Limjunyawong N, Gour N, Pundir P, Dong X. A mast-cell-specific receptor mediates neurogenic inflammation and pain. Neuron. 2019;101:412–420.e3.
Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002.
Ibrahim MZM. The mast cells of the mammalian central nervous system: Part 1. Morphology, distribution and histochemistry. J Neurol Sci. 1974;21:431–78.
Rozniecki JJ, Dimitriadou V, Lambracht-Hall M, Pang X, Theoharides TC. Morphological and functional demonstration of rat dura mater mast cell–neuron interactions in vitro and in vivo. Brain Res. 1999;849:1–15.
Silver R, Silverman AJ, Vitković L, Lederhendler II. Mast cells in the brain: evidence and functional significance. Trends Neurosci. 1996;19:25–31.
Li Z, Liu S, Xu J, Zhang X, Han D, Liu J, et al. Adult connective tissue-resident mast cells originate from late erythro-myeloid progenitors. Immunity. 2018;49:640–653.e5.
Dwyer DF, Barrett NA, Austen KF. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol. 2016;17:878–87.
Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An interleukin-33-mast cell-interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. 2015;43:175–86.
Gessner A, Mohrs K, Mohrs M. Mast cells, basophils, and eosinophils acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are sufficient for rapid cytokine production1. J Immunol. 2005;174:1063–72.
Neumann J. Ueber das Vorkommen der sogenannten „Mastzellen” bei pathologischen Veränderungen des Gehirns. Archiv Pathol Anat Physiol klin Med. 1890;122:378–80.
Rozniecki JJ, Hauser SL, Stein M, Lincoln R, Theoharides TC. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann Neurol. 1995;37:63–6.
Ibrahim MZM, Reder AT, Lawand R, Takash W, Sallouh-Khatib S. The mast cells of the multiple sclerosis brain. J Neuroimmunol. 1996;70:131–8.
Pang X, Letourneau R, Rozniecki JJ, Wang L, Theoharides TC. Definitive characterization of rat hypothalamic mast cells. Neuroscience. 1996;73:889–902.
Shanas U, Bhasin R, Sutherland AK, Silverman AJ, Silver R. Brain mast cells lack the c-kit receptor: immunocytochemical evidence. J Neuroimmunol. 1998;90:207–11.
Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191:813–22.
Tanzola MB, Robbie-Ryan M, Gutekunst CA, Brown MA. Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course 1. J Immunol. 2003;171:4385–91.
Sayed BA, Christy AL, Walker ME, Brown MA. Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: a role for neutrophil recruitment? J Immunol. 2010;184:6891–900.
Xu H, Shi X, Li X, Zou J, Zhou C, Liu W, et al. Neurotransmitter and neuropeptide regulation of mast cell function: a systematic review. J Neuroinflammation. 2020;17:356.
Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM. Mast cell degranulation activates a pain pathway underlying migraine headache. PAIN 2007;130:166.
Nautiyal KM, Ribeiro AC, Pfaff DW, Silver R. Brain mast cells link the immune system to anxiety-like behavior. Proc Natl Acad Sci USA. 2008;105:18053–7.
Hendrix S, Kramer P, Pehl D, Warnke K, Boato F, Nelissen S, et al. Mast cells protect from post-traumatic brain inflammation by the mast cell-specific chymase mouse mast cell protease-4. FASEB J. 2013;27:920–9.
Nelissen S, Vangansewinkel T, Geurts N, Geboes L, Lemmens E, Vidal PM, et al. Mast cells protect from post-traumatic spinal cord damage in mice by degrading inflammation-associated cytokines via mouse mast cell protease 4. Neurobiol Dis. 2014;62:260–72.
Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity. 2012;37:13–24.
Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity. 2011;35:832–44.
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210:535–49.
Karagiannis F, Masouleh SK, Wunderling K, Surendar J, Schmitt V, Kazakov A, et al. Lipid-droplet formation drives pathogenic group 2 innate lymphoid cells in airway inflammation. Immunity 2020;52:620–634.e6.
Wallrapp A, Burkett PR, Riesenfeld SJ, Kim SJ, Christian E, Abdulnour REE, et al. Calcitonin gene-related peptide negatively regulates alarmin-driven type 2 innate lymphoid cell responses. Immunity. 2019;51:709–723.e6.
Tsou AM, Yano H, Parkhurst CN, Mahlakõiv T, Chu C, Zhang W, et al. Neuropeptide regulation of non-redundant ILC2 responses at barrier surfaces. Nature. 2022;611:787–93.
Golebski K, Layhadi JA, Sahiner U, Steveling-Klein EH, Lenormand MM, Li RCY, et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity. 2021;54:291–307.e7.
Mjösberg J, Rao A. Lung inflammation originating in the gut. Science. 2018;359:36–7.
von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 2016;529:221–5.
Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351:1329–33.
Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. 2016;529:226–30.
Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193:3717–25.
Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. 2014;40:758–71.
Sudo T, Motomura Y, Okuzaki D, Hasegawa T, Yokota T, Kikuta J, et al. Group 2 innate lymphoid cells support hematopoietic recovery under stress conditions. J Exp Med. 2021;218:e20200817.
Cardoso V, Chesné J, Ribeiro H, García-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. 2017;549:277–81.
Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. 2017;549:282–6.
Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β2-adrenergic receptor–mediated negative regulation of group 2 innate lymphoid cell responses. Science. 2018;359:1056–61.
Inclan-Rico JM, Ponessa JJ, Valero-Pacheco N, Hernandez CM, Sy CB, Lemenze AD, et al. Basophils prime group 2 innate lymphoid cells for neuropeptide-mediated inhibition. Nat Immunol. 2020;21:1181–93.
Nagashima H, Mahlakõiv T, Shih HY, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP limits group 2 innate lymphoid cell responses and constrains type 2 inflammation. Immunity. 2019;51:682–695.e6.
Pinho-Ribeiro FA, Deng L, Neel DV, Erdogan O, Basu H, Yang D, et al. Bacteria hijack a meningeal neuroimmune axis to facilitate brain invasion. Nature. 2023. https://pubmed.ncbi.nlm.nih.gov/36859544/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1vICqKETsACsk9wtdnzv-f8ZmhLzbpENzbeB4RjIb5torr0Qed&fc=None&ff=20230302044728&v=2.17.9.post6+86293ac.
Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity. 2019;50:707–722.e6.
Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol. 2018;19:1093–9.
Fung ITH, Sankar P, Zhang Y, Robison LS, Zhao X, D’Souza SS, et al. Activation of group 2 innate lymphoid cells alleviates aging-associated cognitive decline. J Exp Med. 2020;217:e20190915.
Russi AE, Walker-Caulfield ME, Ebel ME, Brown MA. Cutting edge: c-Kit signaling differentially regulates type 2 innate lymphoid cell accumulation and susceptibility to central nervous system demyelination in male and female SJL mice. J Immunol. 2015;194:5609–13.
Briukhovetska D, Dörr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21:481–99.
Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21:799–820.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102.
Suzuki A, Leland P, Joshi BH, Puri RK. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine. 2015;75:79–88.
Põlajeva J, Sjösten AM, Lager N, Kastemar M, Waern I, Alafuzoff I, et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS ONE. 2011;6:e25222.
Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. 2012;189:4213–9.
Ruan Z, Zhao Y, Yan L, Chen H, Fan W, Chen J, et al. Single nucleotide polymorphisms in IL-4Ra, IL-13 and STAT6 genes occurs in brain glioma. Front Biosci (Elite Ed). 2011;3:33–45.
Schwartzbaum J, Ahlbom A, Malmer B, Lönn S, Brookes AJ, Doss H, et al. Polymorphisms associated with asthma are inversely related to glioblastoma multiforme. Cancer Res. 2005;65:6459–65.
Demehri S, Cunningham TJ, Manivasagam S, Ngo KH, Tuchayi SM, Reddy R, et al. Thymic stromal lymphopoietin blocks early stages of breast carcinogenesis. J Clin Investig. 2016;126:1458–70.
Mattes J, Hulett M, Xie W, Hogan S, Rothenberg ME, Foster P, et al. Immunotherapy of cytotoxic T cell–resistant tumors by T Helper 2 cells: an eotaxin and STAT6-dependent process. J Exp Med. 2003;197:387–93.
Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Moore EJ, Pierz DM, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995;76:840–52.
Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257:548–51.
Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, et al. Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature 2020;587:121–5.
Harris N, Gause WC. To B or not to B: B cells and the Th2-type immune response to helminths. Trends Immunol. 2011;32:80–8.
Crawford G, Hayes MD, Seoane RC, Ward S, Dalessandri T, Lai C, et al. Epithelial damage and tissue γδ T cells promote a unique tumor-protective IgE response. Nat Immunol. 2018;19:859–70.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med. 2006;354:2689–95.
Schmidts A, Srivastava AA, Ramapriyan R, Bailey SR, Bouffard AA, Cahill DP, et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Rα2 are effective against heterogeneous glioblastoma. Neuro-Oncol Adv. 2023;5:vdac185.
Song E, Mao T, Dong H, Boisserand LSB, Antila S, Bosenberg M, et al. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature. 2020;577:689–94.
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Investig. 2010;120:694–705.
Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science. 2021;373:eabf7844.
Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21:1209–17.
Cai R, Pan C, Ghasemigharagoz A, Todorov MI, Förstera B, Zhao S, et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull–meninges connections. Nat Neurosci. 2019;22:317–27.
Yao H, Price TT, Cantelli G, Ngo B, Warner MJ, Olivere L, et al. Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature. 2018;560:55–60.
Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science. 2021;373:eabf9277.
Mazzitelli JA, Smyth LCD, Cross KA, Dykstra T, Sun J, Du S, et al. Cerebrospinal fluid regulates skull bone marrow niches via direct access through dural channels. Nat Neurosci. 2022;25:555–60.
Pulous FE, Cruz-Hernández JC, Yang C, Kaya Ζ, Paccalet A, Wojtkiewicz G, et al. Cerebrospinal fluid can exit into the skull bone marrow and instruct cranial hematopoiesis in mice with bacterial meningitis. Nat Neurosci. 2022;25:567–76.
Ringstad G, Eide PK. Molecular trans-dural efflux to skull bone marrow in humans with CSF disorders. Brain 2022;145:1464–72.
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7.
Nguyen KD, Qiu Y, Cui X, Sharon Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104–8. https://pubmed.ncbi.nlm.nih.gov/22101429/.
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20.
Morais LH, Schreiber HL, Mazmanian SK. The gut microbiota–brain axis in behaviour and brain disorders. Nat Rev Microbiol. 2021;19:241–55.
Goldman DH, Dykstra T, Smirnov I, Blackburn SM, Mesquita SD, Kipnis J, et al. Age-associated suppression of exploratory activity during sickness is linked to meningeal lymphatic dysfunction and microglia activation. Nat Aging. 2022;2:704–13.
Magistris Lde, Familiari V, Pascotto A, Sapone A, Frolli A, Iardino P, et al. Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J Pediatr Gastroenterol Nutr. 2010;51:418–24. https://doi.org/10.1097/mpg.0b013e3181dcc4a5.
Jyonouchi H. Autism spectrum disorders and allergy: observation from a pediatric allergy/immunology clinic. Expert Rev Clin Immunology. 2010;6:397–411.
Croen LA, Qian Y, Ashwood P, Daniels JL, Fallin D, Schendel D, et al. Family history of immune conditions and autism spectrum and developmental disorders: findings from the study to explore early development. Autism Res. 2019;12:123–35.
Gurney JG, McPheeters ML, Davis MM. Parental report of health conditions and health care use among children with and without autism: National Survey of Children’s Health. Arch Pediatr Adolesc Med. 2006;160:825–30.
Aamodt AH, Stovner LJ, Langhammer A, Hagen K, Zwart JA. Is headache related to asthma, hay fever, and chronic bronchitis? The head-HUNT study. Headache J Head Face Pain. 2007;47:204–12.
Silverberg JI. Comorbidities and the impact of atopic dermatitis. Ann Allergy, Asthma Immunol. 2019;123:144–51.
Acknowledgements
We thank all the members of the Kipnis laboratory for their valuable comments and Shirley Smith for editing the manuscript.
Author information
Authors and Affiliations
Contributions
TM and JK discussed the content of the manuscript, wrote and edited the manuscript, and generated figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mamuladze, T., Kipnis, J. Type 2 immunity in the brain and brain borders. Cell Mol Immunol 20, 1290–1299 (2023). https://doi.org/10.1038/s41423-023-01043-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-023-01043-8
Keywords
This article is cited by
-
Neuroimmunology: reviews and perspectives on recent advances
Cellular & Molecular Immunology (2023)
