
Abstract
Hybridization and introgression, contrary to previous beliefs, are now considered to be widespread processes even among animal species. Nonetheless, the range of their possible outcomes and roles in moulding biodiversity patterns are still far from being fully appraised. Here we investigated the pattern of hybridization and introgression between Salamandrina perspicillata and S. terdigitata, two salamanders endemic to the Italian peninsula. Using a set of diagnostic or differentiated genetic markers (9 nuclear and 1 mitochondrial), we documented extensive unidirectional introgression of S. terdigitata alleles into the S. perspicillata gene pool in central Italy, indicating that barriers against hybridization were permeable when they came into secondary contact and despite their ancient divergence. Nonetheless, purebred S. terdigitata, as well as F1, F2 and backcrosses were not found within the hybrid zone. Moreover, Bayesian analyses of population structure identified admixed populations belonging to a differentiated gene pool with respect to both parental populations. Overall, the observed genetic structure, together with their geographic pattern of distribution, suggests that Salamandrina populations in central Italy could have entered a distinct evolutionary pathway. How far they have gone along this pathway will deserve future investigation.
Similar content being viewed by others
Introduction
Natural hybridization, defined as reproduction between species or divergent populations producing individuals of mixed ancestry1, has long attracted the interest of naturalists (Linnaeus, 1760 cited by2,3; see4 for an historical perspective), yet whether it is a side-effect or a creative evolutionary process is still a matter of debate4,5,6,7,8,9,10,11,12. Once regarded as an incidental and rather ineffective process (e.g.13,14,15), there is now compelling evidence that a large amount of both plant and animal species hybridize (about 10–30%; reviews in4,16), with a diversity of outcomes encompassing reinforcement of pre-mating isolation mechanisms, fusion of hybridizing lineages, hybrid speciation and even adaptive radiations17,18,19,20.
Introgression, which is the invasion of foreign genetic material into a genome16, is a frequent albeit long underappreciated21 outcome of hybridization and a main driver for many of its major evolutionary consequences4,12. The nature and extent of introgression has been shown to substantially vary across interacting lineages. Introgression can be neutral or adaptive, geographically restricted to the contact zone or widespread and transient or persistent; this process can reverse or accelerate the course of speciation events and can also drive populations to follow independent evolutionary pathways12,21.
Hybridization and introgression have been classically studied in natural hybrid zones where two previously allopatric lineages come into secondary contact22, although substantial contributions in this direction have recently emerged from the study of biological invasions23. A major realization coming from the extensive studies of hybrid zones is that substantial differences often occur in the extent of introgression among genomic regions. First, organelle and nuclear genomes commonly differ in the extent of introgression24, often as a consequence of ‘Haldane's rule’, which predict that heterogametic offspring (either XY or ZW) are less viable25,26. Second, there is growing evidence that variation occurs in the extent of introgression even among distinct regions of the nuclear genomes27. These observations provided strong support for the genic view of speciation28, which suggests that reproductive isolation is a consequence of the divergent selection acting on a few loci that are important for fitness and adaptation (‘barrier loci’) rather than to incompatibility between interacting genomes as a whole12,27,28. Accordingly, most of the genome can undergo substantial introgression, whereas genomic regions that are responsible or linked to reproductive or adaptive differences will experience little introgression and will show substantial divergence among hybridizing lineages (the so called ‘islands of genomic divergence’12,27; but see also29). In turn, this view explains why several species remain cohesive evolutionary units while showing clear evidence of extensive introgression among them.
In this paper, we investigated the hybridization dynamics between the only two extant species of salamanders of the genus Salamandrina, the Northern spectacled salamander S. perspicillata and the Southern spectacled salamander S. terdigitata. These species are endemic to the northern and central portion and to the southern portion, of the Italian peninsula, respectively. There is a limited area of close contiguity in-between (see Figure 1). They are lungless and are mainly found in the undergrowth close to various slow running or small lenthic water bodies at altitudes ranging 200–700 m above sea level along the Apennine chain and some adjacent hilly areas30. Once regarded as a single species (S. terdigitata), they were recently identified as two deeply divergent species based on both nuclear and mitochondrial genetic data31,32,33 and their divergence time was estimated to largely predate the onset of the Pleistocene epoch32,33 (but see also34). Recently, a small area was found where both the highly divergent mitochondrial DNA (mtDNA) lineages come into sympatry35 and a preliminary analysis of the hybridization was carried out (based on one sampling location, one mtDNA marker and one diagnostic and one uninformative nuclear markers)36.

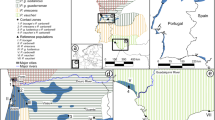
Geographic distribution of Salamandrina perspicillata and S. terdigitata and geographic location of the 10 samples studied.
The map was drawn using the software Canvas 11 (ACD Systems of America, Inc.).
The purpose of this study was to determine the extent, outcome and consequences of hybridization between S. perspicillata and S. terdigitata. We characterized patterns of hybridization and introgression within the putative area of secondary contact, using 9 nuclear (allozyme loci) and 1 mitochondrial markers, whose patterns of variation among allopatric populations have been assessed in previous studies31,32.
Results
Allele frequencies at the 9 loci analysed in the 10 population samples are shown in Table 1. Alleles previously identified to be of diagnostic value for S. perspicillata were not observed in samples 9 and 10, whereas they were frequently prevalent among samples 1–8. Among the five fully diagnostic loci (Pgm-2, Gapdh, Aat-1 PepD-2 and Mdhp-1), alleles of both species were observed at high frequencies in samples 4–8, although S. persipicillata alleles were prevalent in most cases. Significant deviations from the expected Hardy–Weinberg (HW) and genotypic linkage equilibria (at the 5% nominal level) were not observed. Estimates of population genetic variability are presented in Table 1. At all the estimated parameters (HE, HO and AR) samples 4–8 were those showing higher values, whereas samples 9 and 10 were by far the least variable.
The analysis of the genotypes simulated using HYBRIDLAB indicated 0.90 as the threshold value maximizing the confidence in identifying an individual as admixed using STRUCTURE and 0.80 as the best threshold in assigning an individual to a hybrid class using NEWHYBRID. In both cases, the model performance was 0.95 (see Figure 2).

Overall performance to correctly identify individuals simulated by HYBRIDLAB as admixed using STRUCTURE and to assign these to the correct hybrid class with NEWHYBRID, based on five threshold values.
The Bayesian clustering algorithm implemented in STRUCTURE suggested K = 3 as the best clustering option for our data when the highest ln-probability is used as optimality criterion, while K = 2 was indicated as the best option under the Evanno's ΔK optimality criterion (see Supplementary Information). As shown in Figure 3A, with K = 2 all individuals from samples 9 and 10 were attributed to the southern species S. terdigitata, whereas individuals from samples 1–8 were attributed to S. perspicillata. Nevertheless, among samples 4–8 several individuals were identified as significantly admixed with S. terdigitata, with the average Q-value of these samples ranging between 0.8 and 0.15 (see Table 1). When K = 3 was used, samples 9–10 and 1–3 were still assigned to separate clusters, with no evidence of admixture, whereas individuals from samples 4–8 were all assigned to a third cluster (Figure 3B). Among the latter, all but one individuals from samples 5, 6 and 7 appeared admixed with the northern cluster (see also Table 1), whereas no such evidence was observed for individuals from sample 8 and for all but two from sample 4. When populations assigned to each cluster were grouped, the expected heterozygosity (HE) of the northern (samples 1–3), central (4–8) and southern (9–10) clusters was 0.21 (S.D. 0.08), 0.34 (S.D. 0.05) and 0.02 (S.D. 0.02) respectively.

Admixture proportions of each studied individual (barplots) and population sample (pie-diagrams) estimated using STRUCTURE with k = 2 (A) and k = 3 (B), probability of assignment to a particular hybrid class estimated using NEWHYBRID (C) and frequency of S. terdigitata (red) and S. perspicillata (orange) mitotypes within the studied samples (D).
Optimal threshold values estimated with HYBRIDLAB (STRUCTURE: 10–90%; NEWHYBRID: 20–80%) are shown on the barplots as dashed lines. N: pure S. perspicillata; l.c.: pure S. perspicillata assigned with lower confidence; S: pure S. terdigitata; BS: backcross to S. terdigitata; BN: backcross to S. perspicillata; F1 and F2: first and second generation hybrids respectively. The map was drawn using the software Canvas 11 (ACD Systems of America, Inc.).
The analysis of individual genotypic data using NEWHYBRID indicated, with high confidence, that most of the individuals analysed were ‘pure’ S. perspicillata or S. terdigitata; it also showed the lack of F1 and F2 hybrids or backcrosses with S. terdigitata (Figure 3C). However, several individuals from samples 4–8, although showing higher probability of assignment to pure S. perspicillata, did not reach the threshold value (0.80) suggested for an assignment with high confidence.
Finally, the analysis of the mtDNA restriction profiles revealed the occurrence of only two composite haplotypes among the studied individuals, one specific to S. perspicillata and one specific to S. terdigitata (Figure 3D and Table 1). The former was the only one observed among individuals from samples 1–6 and it was also carried by 2 individuals from sample 8, whereas the latter was found fixed in samples 7, 9 and 10 and was prevalent in sample 8 (19 out of 21 individuals analysed).
Discussion
Our analysis of the putative secondary contact zone between S. terdigitata and S. perspicillata highlights the importance of using multiple diagnostic markers in resolving evolutionary processes within such zones, even when studying deeply and anciently divergent species, such as those investigated in the present study. Indeed, the use of the sole mitochondrial genome (as is usual in many barcoding efforts; see e.g.37) would have misleadingly suggested a more northern location for the contact zone and the assignment of most of the individuals from the southern edge of the range of S. perspicillata to S. terdigitata. Furthermore, the high frequencies of several S. terdigitata diagnostic alleles within putatively S. perspicillata samples, together with the occurrence of both HW and linkage equilibria within samples, could have resulted in the misassignment of several individuals to pure S. terdigitata or to a recent hybrid progeny, if each locus had been analysed separately. This could explain, at least in part, the striking discordance between our results and those of previous reports36 that suggested extensive ongoing gene exchange and syntopy between both species based on mtDNA and a single nuclear locus of diagnostic value.
Our data provided evidence of extensive, unidirectional, introgression of the southern species into the northern one. Nevertheless, they provided support neither to the current syntopy between species, nor to ongoing gene exchange, suggesting a more complicated evolutionary scenario for the interaction between the two spectacled salamanders than previously thought.
Diagnostic alleles of S. terdigitata included in the present study were observed to various extents within the southern S. perspicillata populations, whereas the opposite was never observed. A similar pattern of extensive and asymmetric allele sharing among species could be the outcome of either a secondary contact between species followed by hybridization and introgression of one species' genes into the other species' gene pool or of an incomplete sorting of alleles. Nevertheless, we can be fairly confident in discarding incomplete lineage sorting and favoring secondary contact as the causal process of the observed pattern. Indeed, allele sharing was geographically limited to the area of contiguity between both species, not randomly distributed across the species' ranges as expected in the case of incomplete lineage sorting38. The co-presence of both S. perspicillata and S. terdigitata diagnostic alleles within the southern S. perspicillata populations thus serves as evidence of extensive and unidirectional introgression of the southern species' alleles into the northern one's genome.
The frequency of introgressed S. terdigitata alleles within S. perspicillata populations varied conspicuously, ranging from <5% to 55%, with several cases at ≥40% (see Table 1). Additionally, the geographic area where they are found is relatively wide considering the limited dispersal abilities of the studied species30. This is an interesting pattern, suggesting that the various alleles experienced distinct selection regimes once within the heterospecific genomic background27. Nonetheless, this hypothesis deserves further investigation. At the moment, it should be considered as speculative for at least 2 reasons: a) the scattered distribution of both species in the study area and the low number of samples investigated prevented us from comparing clines at each locus with the average extent of introgression (i.e., to undertake a formal genetic cline analysis39,40), as well as to study the role of selection in shaping variation at the studied loci; b) without a more extensive sampling, allowing us to draw geographic trends, we could not discriminate between selection and genetic drift acting on single populations and loci in driving the variation of exogenous allele frequencies over space and time, following secondary contact.
Despite these limitations in our data (which are mostly due to the actual species' distributions, see below), the observed frequencies of several introgressed alleles, as well as the average contribution of S. terdigitata to the genetic diversity of admixed S. perspicillata populations are conspicuous. They appear beyond what we could usually expect for two anciently divergent species with the barriers to gene exchange almost completed at the time of the secondary contact41. Our data suggest that such barriers were leaky, largely permeable when the species came into contact and could have eventually been completed later.
We found no evidence for the occurrence of pure S. terdigitata individuals or recent hybrids (two generations) within southern S. perspicillata populations (see Figure 2C). At least three scenarios could account for such an absence: 1) pure S. terdigitata individuals are present but rare within the study area and our dataset lacks the resolution to reliably identify recent hybrids; 2) our sampling area did not cover the core of the hybrid zone, where both pure parentals and hybrids occur; or 3) pure S. terdigitata are no longer present within the range of S. perspicillata. The analysis of model performance using simulated hybrid genotypes using both STRUCTURE and NEWHYBRID indicated that our data provided the necessary resolution to identify recent hybrids, leading us to discount the first scenario as the least probable. Also, the key question to disentangle scenario 2 and 3 is whether the hybrid zone (and its center) could extend more to the south and east of our sample 8, towards the area where pure S. terdigitata populations thrive. Unfortunately, this question does not have a simple answer. Currently, the distribution of Salamandrina populations is not continuous along the north-west to south-east axis and the geographic gap between samples 8 and 9 largely reflects a discontinuity in the distribution of the populations. However, this area has been intensively modified by past and present anthropogenic activities and it is not implausible that the structure of the hybrid zone has been modified as well. Therefore, while scenario 3 appears to be the most plausible in current times, we cannot exclude that scenario 2 occurred at some point in the past.
Completion of reproductive isolation barriers driven by production of unfit hybrids (i.e., by reinforcement9,18,) followed by exclusion of S. terdigitata from the sympatric area (i.e., scenario 3) on the one hand and the recent disappearance of part of the hybrid zone where the two species met and mated (i.e., scenario 2) on the other hand, could be tested experimentally. Indeed, under scenario 3, experimental investigations of mate choice using S. perspicillata and S. terdigitata individuals from the study area should reveal the occurrence of pre-zygotic barriers (by a strong deficit of heterospecific mates), whereas such barriers could not intervene when individuals from largely allopatric populations are tested42,43. The same outcome would not be expected if scenario 2 were true. Therefore, such an experimental design, based on the theory of reinforcement of reproductive isolation, would use the expected geographic structure of reinforcing selection and its outcomes to generate testable hypotheses12 and to shed light on the history of interactions between the two Salamandrina species. We are currently exploring this research direction.
On a distinct but similar ground, results using STRUCTURE with K = 3 as a clustering option identified the southernmost populations of S. perspicillata as belonging to a differentiated gene pool with respect to those located more to the north. Interestingly, when this clustering option was adopted, the degree to which S. terdigitata contributed to the gene pool of southern S. perspicillata populations appeared negligible, whereas gene flow from the northern populations was indicated. This pattern supports the idea that the S. terdigitata alleles have become integral to the gene pool of southern S. perspicillata populations and it also suggests that these populations could have achieved some degree of evolutionary ‘independence’ from conspecific populations to the north. Further support for this interpretation comes from the lack of HW and linkage disequilibria, as well as from previous findings35 that have indicated that the southern S. perspicillata populations belong to a distinct albeit weakly differentiated mtDNA haplogroup.
To what extent this group of populations has entered its own evolutionary pathway will certainly deserve future investigation using a deeper genome scan, as well as a thorough analysis of trait variations in ecological and morphological characters, both among and within populations. Nonetheless, it is worth noting that the aforementioned genetic pattern shows striking parallels with patterns previously used in support of the hybrid origin of recently originated lineages44,45,46. Regardless of how far these have progressed in this pathway, the observed genetic structure, together with their patchy distribution within a heterogeneous and recently human-disturbed area, render Salamandrina populations in central Italy particularly interesting to contribute to investigations of introgressive hybridization, particularly in terms of its range of outcomes47. In times of resurgent and growing interest in the role of reticulate evolution in shaping current patterns of biodiversity, these appear to offer intriguing opportunities for future insights.
Methods
Sampling and laboratory procedures
Population samples were collected at 10 sites (157 individuals) from the area of close contiguity between the ranges of Salamandrina perspicillata and S. terdigitata. The geographic location of the population samples and sample sizes are shown in Table 2 and Figure 1. For each individual analysed, a tissue sample was obtained through tail-clipping and the individual was released at its collection site. Tissue samples were then transported to the laboratory and stored at −80°C. Sampling activities and the tail-clipping procedure for tissue collection were approved by the Italian Ministry of Environment (permit number: DPN-2009-0026530).
Standard horizontal starch gel (10%) electrophoresis was conducted to screen for variations at nine allozyme loci previously identified as showing diagnostic or differentiated electrophoretic patterns among the two species31. These loci were: Lactate dehydrogenase (Ldh-1; EC 1.1.1.27), Malate dehydrogenase NADP+ (Mdhp-1; EC 1.1.1.40), Isocitrate dehydrogenase (Icdh-2; EC 1.1.1.42), Glyceraldehyde-3-phosphate dehydrogenase (Gapdh; EC1.2.1.12), Aspartate transaminase (Aat-1; EC 2.6.1.1), L-phenylalanyl-L-proline peptidase (Pep-D2; EC 3.4.13.9), Mannose phosphate isomerase (Mpi; EC5.3.1.8), Glucose phosphate isomerase (Gpi; EC 5.3.1.9) and Phosphoglucomutase (Pgm-2; EC 5.4.2.2). Allozyme electrophoresis, zymogram visualization and allele calling procedures were carried out following previously published protocols31.
Genomic DNA was extracted using the cetyltrimethyl ammonium bromide (CTAB) procedure48. A fragment of the mitochondrial DNA (mtDNA) gene encoding for cytochrome b was amplified through polymerase chain reaction (PCR) and sequenced. The PCR mixture and cycling conditions followed strictly32. PCR products of two individuals per population sample were purified and sequenced by Macrogen Inc. (www.macrogen.com). These sequences were then checked and aligned using the software GeneStudio Pro and used to identify two restriction endonucleases of diagnostic value among S. terdigitata and S. perspicillata. The many putative restriction enzymes were further assessed for their diagnostic value using previously published sequences of both species, available in the Genbank database. The enzymes SspI and AluI were selected for the assessment of the restriction fragment length polymorphisms (RFLP) among all the individuals used in the present study. For this purpose, 10 μl of each PCR product was digested overnight with five units of enzyme, following manufacturer's instructions (Promega Corporation). Restriction fragments were separated on 3% agarose gels, stained with GelRed (Biotium) and visualized under UV light.
Data analysis
Basic descriptive statistics of the allozyme dataset were computed using the softwares FSTAT 2.9.3 and BIOSYS-2. These included population allele frequencies, observed (HO) and unbiased expected (HE) heterozygosity and allelic richness (AR, an estimate of the average number of alleles per locus corrected for sample size). FSTAT was also used to test departures from the expected Hardy–Weinberg (HW) equilibrium and genotypic linkage equilibrium between pairs of loci in each population sample.
The analysis of the occurrence and extent of admixture between the two spectacled salamanders within their putative area of secondary contact was conducted using two methodological approaches: the Bayesian clustering algorithm implemented in the software STRUCTURE 2.3.449 and the Bayesian analysis of the genotypic classes (pure, F1, F2 and backcrosses) as implemented in NEWHYBRID50.
The analysis with STRUCTURE was conducted using a model allowing for admixture and independent allele frequencies among populations. Given the main purpose of this study, we were particularly interested in a model with two clusters (i.e., K = 2), to assess the occurrence of individuals of mixed ancestry in our sample. Nevertheless, to explore the occurrence of further population structure within both species we ran STRUCTURE with K ranging from 1 to 10 and we analyzed results both with K = 2 and with the best clustering option, as suggested by the post-processing of the STRUCTURE output. For each value of K we carried out 10 replicates of the analysis, with 100,000 Markov Chain Monte Carlo (MCMC) iterations following a burn-in of 50,000 iterations, as these settings guaranteed convergence of the Markov chains to a stationary distribution (see Supplementary Information). The results of the analysis using STRUCTURE were summarized and analysed using STRUCTURE HARVESTER51.
The assignment of individuals to the various hybrid classes with NEWHYBRID was performed by computing 100,000 MCMC iterations following 20,000 iterations discarded as burn-in (after checking for stationarity). The analysis was run with two population samples (which received a q > 0.95 during previous STRUCTURE runs) pre-assigned as parental (i.e., ‘z’ option in use), following suggestions by50.
The best threshold values to confidently identify an individual as admixed in the STRUCTURE analyses, or to assign it to a particular hybrid class in NEWHYBRID, were identified using the approach of52. We selected 30 individuals receiving q > 0.95 during preliminary STRUCTURE runs and used them to simulate 100 individuals of each hybrid class (pure S. terdigitata, pure S. perspicillata, F1, F2, backcross to S. terdigitata and backcross to S. perspicillata) using the program HYBRIDLAB 1.053. This program generates hybrid genotypes by randomly sampling alleles at each locus as a function of the respective frequencies and assuming random mating, linkage equilibrium and markers' neutrality. No specific parameter settings are allowed by HYBRIDLAB. We repeated this procedure 10 times and ran the analyses with both STRUCTURE and NEWHYBRID using the same settings employed for the real dataset. Results based on simulated genotypes were used to estimate efficiency (the proportion of individuals in a group that were correctly identified), accuracy (the proportion of an identified group that truly belongs to that category) and performance (the product of efficiency and accuracy, varying from 0 [min] to 1 [max]) of the two methods under the threshold values 0.95, 0.90, 0.85, 0.80 and 0.75. Finally, for each method, the threshold value maximizing the overall performance of the model was retained and used to analyse the real dataset.
References
Barton, N. H. & Hewitt, G. M. Analysis of Hybrid Zones. Annu. Rev. Ecol. Evol. Syst. 16, 113–148 (1985).
Grant, V. Plant Speciation, 2nd edn (Columbia University Press, New York, 1981).
Darwin, C. On the origin of species (John Murray, London, 1859).
Arnold, M. L. Evolution Through Genetic Exchange (Oxford University Press, Oxford, 2006).
Stebbins, G. L., Jr The role of hybridization in evolution. Proc. Am. Philos. Soc. 103, 231–251 (1959).
Dowling, T. E. & Secor, C. L. The role of hybridization and introgression in the diversification of animals. Annu. Rev. Ecol. Syst. 28, 593–619 (1997).
Schemske, D. W. Understanding the origin of species. Evolution. 54, 1069–1073 (2000).
Barton, N. H. The role of hybridization in evolution. Mol. Ecol. 10, 551–568 (2001).
Coyne, J. A. & Orr, H. A. Speciation. (Sinauer Associates, Sunderland, MA, 2004).
Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20, 229–237 (2005).
Soltis, P. S. & Soltis, D. E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588 (2009).
Abbott, R. et al. Hybridization and speciation. J. Evol. Biol. 26, 229–246 (2013).
Dobzhansky, T. Speciation as a stage in evolutionary divergence. Am. Nat. 74, 312–321 (1940).
Mayr, E. Systematics and the origin of species. (Columbia University Press, New York, 1942).
Mayr, E. Animal Species and Evolution. (Cambridge, Mass., Harvard University Press, 1963).
Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20, 229–237 (2005).
Seehausen, O. Hybridization and adaptive radiation. Trends Ecol. Evol. 19, 198–207 (2004).
Servedio, M. R. & Noor, M. A. F. The role of reinforcement in speciation: theory and data. Annu. Rev. Ecol. Syst. 34, 339–364 (2003).
Mallet, J. Hybrid speciation. Nature. 446, 279–283 (2007).
Seehausen, O., van Alphen, J. J. M. & Witte, F. Cichlid fish diversity threatened by eutrophication that curbs sexual selection. Science. 277, 1808–1811 (1997).
Rheindt, F. E. & Edwards, S. Genetic introgression: an integral but neglected component of speciation in birds. Auk. 128, 620–632 (2011).
Hewitt, G. M. Quaternary phylogeography: the roots of hybrid zones. Genetica. 139, 617–638 (2011).
Fitzpatrick, B. M. et al. Rapid fixation of non-native alleles revealed by genome-wide SNP analysis of hybrid tiger salamanders. BMC Evol. Biol. 9, 176 (2009).
Toews, D. P. L. & Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930 (2012).
Haldane, J. B. S. Sex ratio and unisexual sterility in hybrid animals. J. Genet. 72, 101–109 (1922).
Wu, C. I., Johnson, N. A. & Palopoli, M. F. Haldane's rule and its legacy: Why are there so many sterile males? Trends Ecol. Evol. 11, 281–284 (1996).
Seehausen, O. et al. Genomics and the origin of species. Nat. Rev. Genet. 15, 176–192 (2014).
Wu, C.-I. The genic view of the process of speciation. J. Evol. Biol. 14, 851–865 (2001).
Cruickshank, T. E. & Hahn, M. W. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157 (2014).
Lanza, B., Nistri, A. & Vanni, S. Iconography of Italian Amphibians. (Grandi & Grandi Editori, Savignano sul Panaro, Modena, 2006).
Canestrelli, D., Zangari, F. & Nascetti, G. Genetic evidence for two distinct species within the Italian endemic Salamandrina terdigitata Bonnaterre, 1789 (Amphibia: Urodela: Salamandridae). Herpetol. J. 16, 221–227 (2006).
Nascetti, G., Zangari, F. & Canestrelli, D. The spectacled salamanders, Salamandrina terdigitata, Lacepede, 1788 and S. perspicillata Savi, 1821: 1) genetic differentiation and evolutionary history. Rend. Fis. Acc. Lincei. 16, 159–169 (2005).
Mattoccia, M., Romano, A. & Sbordoni, V. Mitochondrial DNA sequence analysis of the spectacled salamander, Salamandrina terdigitata (Urodela: Salamandridae), supports the existence of two distinct species. Zootaxa. 995, 1–19 (2005).
Hauswaldt, J. S. From species divergence to population structure: A multimarker approach on the most basal lineage of Salamandridae, the spectacled salamanders (genus Salamandrina) from Italy. Mol. Phylogenet. Evol. 70, 1–12 (2014).
Mattoccia, M., Marta, S., Romano, A. & Sbordoni, V. Phylogeography of an Italian endemic salamander (genus Salamandrina): glacial refugia, postglacial expansions and secondary contact. Biol. J. Linn. Soc. 104, 903–992 (2011).
Hauswaldt, J. S., Angelini, C., Pollok, A. & Steinfartz, S. Hybridization of two ancient salamander lineages: molecular evidence for endemic spectacled salamanders on the Apennine Peninsula. J. Zool. 284, 248–256 (2011).
Kress, W. J. & Erickson, D. L. DNA barcodes: methods and protocols. (Methods in Molecular Biology, 858, Springer, Berlin, 2012).
Avise, J. C. Molecular markers, natural history and evolution. 2nd edition. (Sinauer Associates, Sunderland, Massachusetts, 2004).
Szymura, J. M. & Barton, N. H. Genetic analysis of a hybrid zone between the fire-bellied toads near Cracow in southern Poland. Evolution. 40, 1141–1159 (1986).
Gompert, Z. & Buerkle, C. A. A powerful regression based method for admixture mapping of isolation across the genome of hybrids. Mol. Ecol. 18, 1207–1224 (2009).
Gompert, Z., Parchman, T. L. & Buerkle, C. A. Genomics of isolation in hybrids. Phil. Trans. R. Soc. B. 367, 439–450 (2012).
Urbanelli, S. & Porretta, D. Evidence of reinforcement of premating isolation between two species of the genus Ochthebius Coleoptera: Hydraenidae. Evolution. 62, 1520–1527 (2008).
Porretta, D., Mastrantonio, V., Bellini, R., Somboon, P. & Urbanelli, S. Glacial History of a Modern Invader: Phylogeography and Species Distribution Modelling of the Asian Tiger Mosquito Aedes albopictus. PLoS ONE. 7, e44515. 10.1371/journal.pone.0044515 (2012).
Gompert, Z., Fordyce, J. A., Forister, M., Shapiro, A. M. & Nice, C. C. Homoploid hybrid speciation in an extreme habitat. Science. 314, 1923–1925 (2006).
Gompert, Z. et al. Bayesian analysis of molecular variance in pyrosequences quantifies population genetic structure across the genome of Lycaeides butterflies. Mol. Ecol. 19, 2455–2473 (2010).
Brelsford, A., Mila, B. & Irwin, D. E. Hybrid origin of Audubon's warbler. Mol. Ecol. 20, 2380–2389 (2011).
Nolte, A. W. & Tautz, D. Understanding the onset of hybrid speciation. Trends Genet. 26, 54–58 (2010).
Doyle, J. & Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 1115 (1987).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics. 155, 945–959 (2000).
Anderson, E. C. & Thompson, E. A. A model-based method for identifying species hybrids using multilocus genetic data. Genetics. 160, 1217–1229 (2002).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Vähä, J.-P. & Primmer, C. R. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 15, 63–72 (2006).
Nielsen, E. E. G., Arvebach, L. & Kotlicki, P. HYBRIDLAB (version 1.0): a program for generating simulated hybrids from population samples. Mol. Ecol. Notes. 6, 971–973 (2006).
Acknowledgements
We are grateful to Pietro Bagalà and Francesco Spallone for their help with sample collection and to Daniele Porretta and Francesca Zangari for their kind discussions during the early phases of manuscript preparation. The Italian Ministry of Education, University and Research (PRIN project 2012FRHYRA) funded this study.
Author information
Authors and Affiliations
Contributions
D.C. and G.N. conceived and designed the study. D.C., R.B. and G.N. performed the experiments. D.C. and R.B. analysed the data. D.C. and R.B. wrote the paper. All authors reviewed and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Informations
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Canestrelli, D., Bisconti, R. & Nascetti, G. Extensive unidirectional introgression between two salamander lineages of ancient divergence and its evolutionary implications. Sci Rep 4, 6516 (2014). https://doi.org/10.1038/srep06516
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06516
This article is cited by
-
Ancient hybridization and mtDNA introgression behind current paternal leakage and heteroplasmy in hybrid zones
Scientific Reports (2019)
-
Evidences for a shared history for spectacled salamanders, haplotypes and climate
Scientific Reports (2018)
-
Hybridization and extensive mitochondrial introgression among fire salamanders in peninsular Italy
Scientific Reports (2018)
-
An Arabidopsis introgression zone studied at high spatio-temporal resolution: interglacial and multiple genetic contact exemplified using whole nuclear and plastid genomes
BMC Genomics (2017)
-
A combination of long term fragmentation and glacial persistence drove the evolutionary history of the Italian wall lizard Podarcis siculus
BMC Evolutionary Biology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
