
Abstract
Population genetic studies provide accurate information on population structure, connectivity, and hybridization. These are key elements to identify units for conservation and define wildlife management strategies aimed to maintain and restore biodiversity. The Mediterranean island of Sardinia hosts one of the last autochthonous mouflon populations, descending from the wild Neolithic ancestor. The first mouflon arrived in Sardinia ~ 7000 years ago and thrived across the island until the twentieth century, when anthropogenic factors led to population fragmentation. We analysed the three main allopatric Sardinian mouflon sub-populations, namely: the native sub-populations of Montes Forest and Mount Tonneri, and the reintroduced sub-population of Mount Lerno. We investigated the spatial genetic structure of the Sardinian mouflon based on the parallel analysis of 14 highly polymorphic microsatellite loci and mitochondrial D-loop sequences. The Montes Forest sub-population was found to harbour the ancestral haplotype in the phylogeny of European mouflon. We detected high levels of relatedness in all the sub-populations and a mitochondrial signature of hybridization between the Mount Lerno sub-population and domestic sheep. Our findings provide useful insights to protect such an invaluable genetic heritage from the risk of genetic depletion by promoting controlled inter-population exchange and drawing informed repopulation plans sourcing from genetically pure mouflon stocks.
Similar content being viewed by others

Introduction
Ovine domestication began in the Fertile Crescent around 10,000 years ago (YA) from mouflon-like animals with horns and short-shedding wool1,2,3. A second wave of domestication occurred 6000 years later and led to the modern sheep: polled with no-shedding wool4,5. As a consequence, the rearing of the first mouflon-like ovine was abandoned in favour of the new economically valuable one6. The original mouflon-like went back to feral life but gradually disappeared in mainland Europe due to intense hunting and habitat erosion. However, relics of these ancestral populations survived in the harshest mountainous areas of the Mediterranean islands of Sardinia, Corsica, and Cyprus, where they established populations which survived until now4,5,7,8. Since the eighteenth century, Corsican and Sardinian mouflon have been used to repopulate several regions of mainland Europe. During the last century, the Corsican and Sardinian mouflon populations experienced severe size contractions due to over-hunting and habitat erosion. However, the implementation of conservation policies in the last 50 years brought the populations to increase and reach the current number of ~1000 and ~6000 individuals in Corsica and Sardinia, respectively9,10,11.
The most credited hypothesis dated the arrival of mouflon in Sardinia around 7000 YA7,8,12,13, followed ~3–4000 years later by the ovines of the second wave of domestication. In the twentieth century the competition for grazing land between mouflon and domestic sheep, the intensified hunting pressure, and the more recent use of barbed-wire fences along the roads strongly altered the mouflon natural habitat, resulting in population fragmentation into small, isolated groups often confined to less favourable habitats14. Finally, the outbreak of newly introduced infectious diseases (e.g., the blue tongue) contributed to further reduce the mouflon population size15.
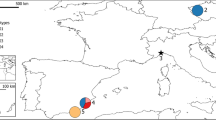
Currently, the mouflon distribution is limited to eastern Sardinia (Ogliastra, Gennargentu and Supramonte), including the areas of Mount Tonneri (Seui), Montes Forest (Orgosolo), and Mount Albo (Lula), and a few managed and protected areas where mouflon has been reintroduced. This range covers only a limited part of the original species distribution16 (Fig. 1). In 1998, the Sardinian Regional Council for Environment started an action plan to increase the number and distribution of mouflon across Sardinia by transferring small groups of mouflons sourced from the historical mouflon range into enclosures before being released into new areas (Legge Regionale n 26). Population bottlenecks across several generations can lead small, isolated populations to rapidly lose genetic diversity as a result of genetic drift17,18,19, at a speed that depends on the severity of the bottleneck and the standing diversity of the founder population20,21,22. Further, in the case of the Sardinian mouflon, hybridization with domestic sheep may affect the genetic integrity of mouflon through the introgression of maladapted genetic components7. Crossbreeding between mouflon and sheep has been reported since Roman times23 and still occurs24,25. This is often the case for mouflon colonies established for hunting purposes in several European countries, where the practice of human-mediated crossbreeding was used to produce hybrids with increased body size and bigger horns26.

Historical and current distribution of the mouflon in Sardinia and sampling sites. Black dashed lines represent the two main regional connection roads. Red arrows indicate the three steps of translocation of the animals selected for reintroduction in the Mount Lerno area. Map was generated in Inkscape v1.0 (https://inkscape.org/).
A better knowledge of the genetic differentiation of wildlife populations and the improved understanding of the selective pressure acting upon them are essential tools for the effective management and conservation of endangered species. Molecular markers like mitochondrial DNA (mtDNA) and microsatellites are commonly used to evaluate the genetic health of a population and to define conservation units representing valuable reservoir of genetic variation. Studies based on genome-wide markers and whole mtDNA sequence have shown low levels of genetic diversity and the occasional occurrence of domestic sheep introgression signals in the Sardinian mouflon populations5,7,27.
To date, five domestic sheep mitochondrial lineages, defined as haplogroups (HPGs) A, B, C, D, and E, have been identified28. HPG-B is the most common haplogroup among sheep breeds28,29 and is also shared by the mouflon from Sardinia and Corsica. The Sardinian and Corsican mouflon are classified as European mouflon (Ovis gmelini musimon) along with the reintroduced mouflon populations from mainland Europe, despite the great genetic distance detected between the Corsico-Sardinian and the European mouflon mtDNA lineages5,8. The Corsico-Sardinian mouflon clade shows an early split from the evolutionary branch originating the domestic sheep HPG-B and the mainland Europe mouflon lineage5. Further, the Sardinian mouflon gene pool harbours the oldest HPG-B haplotype identified so far8. Such findings suggest the need to revise the systematic classification and nomenclature of these species. Until a new nomenclature is established, in this report we choose to identify the individuals from mainland Europe as European mouflon, whereas we will refer to insular individuals as Sardinian or Corsican mouflon, according to their origin.
Here, we use mtDNA and microsatellite markers to describe the genetic diversity and population structure of the Sardinian mouflon. We estimate the genetic differentiation among and within three representative sub-populations from the historical range of Sardinian mouflon and evaluate the effect of landscape anthropogenic alterations. Finally, we provide the first data on the spatial genetic structure of mouflon in Sardinia.
Results
Mitochondrial DNA
We generated a total of 54 sequences of heterogeneous length ranging from 460 to 1180 bp which were deposited in GenBank (GB# MW727287-340). In order to estimate the genetic variability on the entire available sample, the analyses were carried out on a 439 bp fragment of the hypervariable domain I of the mitochondrial D-loop region. A total of five haplotypes were detected: Hpt-1 (GB# MG489885) previously identified in Sardinia8 and belonging to the HPG-B, Hpts 2–4 described for the first time in this work, and Hpt-5 (GB# KF228720) previously found in Comisana sheep30.
Within the Montes Forest sub-population three haplotypes separated by one-step mutations were observed, resulting in a haplotype diversity of 0.62 (Table 1). Only one private haplotype was found for each of the other two sub-populations. The five haplotypes were separated by seven polymorphic sites and represented an overall haplotype diversity of 0.74 and nucleotide diversity of 0.006 (Table 1).
The components of the total genetic variation were estimated by AMOVA and showed a higher proportion of genetic diversity variation “among populations” (79.3%) than “within population” (20.7%) (Table 2).
The median-joining network (MJN) analysis detected two main D-loop haplotype clusters well separated from the outgroup (Fig. 2). A clear geography-based cluster composition was revealed: Cluster I included sequences of mouflons from Asia and Cyprus, while Cluster II comprised all the sequences from Europe and a few from Asia. The five haplotypes found in Sardinia are represented in Cluster II. The Hpt-1 was positioned in the centre and directly connected to all the other haplotypes.

Median-joining network of mtDNA D-loop haplotypes as inferred from the analysis of 88 Ovis gmelini sequences. Three additional homologous sequences from urial (O. vignei) were included as outgroups. Haplotype clusters (Outgroups—Cluster I–Cluster II) are indicated by dashed squares. The number of mutations between haplotypes is indicated by perpendicular lines. The size of the circles is proportional to the frequency of a certain haplotype in the total sample, with colours assigned by geographical origin. Labels referring to haplotypes detected within Sardinian samples are also reported.
To quantify D-loop divergence within the Sardinian mouflon population (n = 54), pairwise genetic distances between groups were calculated (Supplementary Table S1). The mean divergence was 0.005 ± 0.002 and 0.01 ± 0.004 at the intra- and inter-population level, respectively.
Microsatellites
Fourteen out of sixteen genotyped microsatellite loci were found polymorphic in at least one of the three sub-populations with a mean of four alleles by locus (Supplementary Table S2). Micro-Checker suggested the presence of null allele at the MCM139 locus. This locus was discarded from further analyses. Seven, two and three microsatellite loci deviated from HWE in Montes Forest, Mount Tonneri and Mount Lerno sub-populations, respectively (Supplementary Table S3). No locus deviated from HWE in all of the three sub-populations. The mean allelic richness across loci was higher (ANOVA: P < 0.01) in Montes Forest sub-population (4.17) when compared to Mount Tonneri (2.71) and Mount Lerno (2.54), indicating a higher genetic variability in this sub-population.
FIS estimates were not statistically significant neither at the global nor at sub-population levels, except for the Montes Forest sub-population (Table 3).
The FST values were high both across the three sub-populations (FST = 0.30; 0.218–0.399; P < 0.01) and for pairwise comparisons, revealing a strong genetic differentiation. The highest genetic distance was recorded between Mount Lerno and Mount Tonneri (FST = 0.33; 0.174–0.461; P < 0.01), whereas the Montes Forest sub-population was found to be almost equally differentiated from the other two (vs Mount Lerno: FST = 0.27; 0.184–0.390; P < 0.01 and vs Mount Tonneri: FST = 0.27; 0.190–0.384; P < 0.01).
The AMOVA indicated that the within population variation accounted for 0.66% of total variation, whereas differences among populations contributed for 29.57% of the total variation (Table 2). In addition, most of the variance (69.77%) was partitioned within individuals reflecting a high genetic variation at the individual level.
In the PCA (Fig. 3), the scree plot identified the first three principal components (PC) as the most informative (Supplementary Fig. S1). The first two PC combined accounted for 30.4% of the total variance and identified three distinct groups corresponding to the three sampling sites of Montes Forest, Mount Lerno and Mount Tonneri. One individual from the Mount Tonneri sub-population appeared in an intermediate position between the Mount Tonneri and Montes Forest groups. PC3 accounted for 8.4% of the variance and split the Montes Forest sub-population in two groups.

Principal component analysis of the three Sardinian mouflon sub-populations based on 13 microsatellite markers. The percentage values within brackets refer to the proportion of variance explained by each of the displayed principal components.
Since gene flow and inbreeding are known to affect allele frequencies, we assessed migration rate (Table 4) and relatedness. Data showed a low migration rate among sub-populations with the highest gene flow detected between Mount Tonneri and Montes Forest (2.1% migrants/generation). Lower values were found between Montes Forest and Mount Lerno (mean 1.8%), and between Mount Tonneri and Mount Lerno (mean 1.6%). Most of the individuals in each sub-population derived from their source population (Mount Lerno 96.23%; Mount Tonneri 96.58%; Montes Forest 96.19%).
The median (± SD) values of the r frequency distributions inferred by the relatedness analysis were 0.054 (± 0.258) for Montes Forest, 0.351 (± 0.175) for Mount Lerno and 0.264 (± 0.227) for Mount Tonneri sub-populations.
The mean relatedness between sub-populations was 0.13, 0.33 and 0.37 for Montes Forest, Mount Tonneri and Mount Lerno, respectively (ANOVA: P < 0.01) (Supplementary Table S4). A multivariate analysis of pairwise relatedness distances identified two familial groups within the Montes Forest sub-population. No sub-structure was observed at the mtDNA level since the three haplotypes (Hpts 1–3) were found equally distributed across sub-populations (Supplementary Fig. S2).
As expected, the presence of highly related individuals, either full siblings or parent-offsprings, was observed (r > 0.5; Fig. 4) with no statistical difference among sub-populations (χ2 test: P = 0.245). The proportion of r values > + 0.5 was 13.1%, 19.2% and 19.6% in Montes Forest, Mount Tonneri and Mount Lerno sub-populations, respectively. One individual sourced from Mount Tonneri recorded the highest r estimates of 0.49 and 0.51 with one individual from Montes Forest and one from Mount Tonneri, respectively.

Distribution of pairwise relatedness coefficients showing the presence of highly related individuals in each of the three Sardinian mouflon sub-populations.
A strong signature of genetic bottleneck was detected in the Mount Lerno sub-population as indicated by the shifted mode curve resulting from the allelic frequencies distribution. Moreover, the one-tailed Wilcoxon test evidenced a significant heterozygosity excess for all the three mutation models (IAM, TPM and SMM; P < 0.05) (Table 5).
Our results showed that the effective population size (Ne) was not affected by allele frequencies lower than 0.02. The Montes Forest sub-population showed the highest Ne mean value (2.8) compared to Mount Tonneri (2.4) and Mount Lerno (2.2) (Table 6). The analysis of the 95% CIs differences evidenced overlapping ranges between all sub-populations with the narrowest ranges in Montes Forest (CIs = 2.3–4.0) and Mount Tonneri (CIs = 1.7–3.3) sub-populations with respect to the Mount Lerno one (CIs = 1.4–5.3).
Discussion
Remnant island populations often represent unique genetic reservoirs due to their distinctiveness and should be always prioritised for conservation31,32. To this end, knowledge on their spatial distribution, population structure and connectivity are key to draw informed conservation plans33,34,35. We analysed 14 highly polymorphic microsatellite loci and D-loop sequences to describe the diversity and structure of the current Sardinian mouflon autochthonous population, providing the first evidence of a clear genetic structuring.
Mitochondrial DNA
We detected a total of five mtDNA haplotypes, including Hpt-1 that has been described as the oldest haplotype within the HPG-B8. Among the other four, three were novel (Hpts 2, 3 and 4) while Hpt-5, found in the Mount Lerno population, was previously detected in the Comisana domestic sheep breed30 but never recorded in mouflon. Noticeably, none of the haplotypes detected within Montes Forest and Mount Tonneri sub-populations were found to be shared with domestic sheep. We hypothesize that during the subsequent steps of the translocation process from Mount Albo to Mount Lerno, passing through Capo Figari and the Asinara Island, contact with domestic breeds could have occurred.
Each sampling site showed a unique haplotype composition, suggesting a minimal matrilineal gene flow, in accordance with the high genetic differentiation detected among populations (FST = 0.79).
To position the Sardinian mtDNA haplotypes within a broader evolutionary context, we performed a network analysis including additional sequences of mouflon from Europe and Asia highlighting the presence of two clear phylogeographic clusters, including haplotypes of Asian and European origin, respectively (Fig. 2). Within the European cluster both Sardinian and Corsican mouflon showed private haplotypes, whereas two of the three European mouflon sequences grouped with two Asian mouflons in the same haplotype, as previously reported5,8. We speculate that the European and Asian mouflon individuals carrying this haplotype are likely to be mouflon rams x sheep ewe hybrids erroneously classified as pure mouflons based on morphological traits.
The central position of Hpt-1 in the network suggests its ancestral role in the phylogeny of Ovis. We found this haplotype in the Montes Forest sub-population, which supports it as the historical memory of the wild pool introduced in Sardinia by the first settlers during the Neolithic. However, additional data on Corsican mouflon are necessary to gain a complete view on the arrival of mouflon into the islands of the central Mediterranean area.
Our results highlighted the genetic differences between Corsico-Sardinian and European mouflons also found in previous works5,8, although additional sampling of individuals sourced from different parts of mainland Europe is necessary to confirm this observation. Importantly, the current nomenclature still aggregates Sardinian, Corsican, and mainland European mouflon under the same systematic classification. In 1994 Cugnasse36 suggested to refer to mainland mouflon populations with the scientific name O. g. musimon × Ovis sp. to account for their possibly impaired genetic integrity. Based on our mitochondrial results we believe appropriate to discriminate the Corsico-Sardinian mouflon and the mainland European mouflon as two different sub-species. A similar approach was implemented in the case of the Anatolian (O. g. anatolica) and Cypriot (O. g. ophion) mouflons5,8,37.
Microsatellites
In accordance with the mtDNA-based AMOVA, microsatellite analyses showed high FST values and the presence of private alleles (n = 12), probably due to a reduced gene flow. However, caution is required in interpreting these results due to the small sample sizes available38. Previous studies on microsatellite loci39,40 recommended to sample at least 20–25 individuals to reduce errors and increase accuracy. Nonetheless, the calculation of genetic differentiation indices is still informative despite low sample sizes, as is often the case when studying small, endangered populations.
The mitochondrial/microsatellites FST ratio was > 2, suggesting higher male-biased natal dispersal. This is expected, as mouflon rams are more prone to disperse during rutting phase, albeit mouflon adopt a polygynous reproductive system with philopatric males and females22,41,42.
High levels of relatedness were recorded in all sub-populations likely due to inbreeding (Fig. 4). The polygynous mating behaviour of mouflon might also justify these levels of relatedness, due to restricted dispersal and the consequent increased relatedness among potential mates43,44. Furthermore, high relatedness is favoured in small populations where the low number of individuals increases the probability of mating with close relatives.
The PCA and FST results showed a clear differentiation among the three sub-populations. The presence of two groups detected in Montes Forest sub-population both by PCA and multivariate analysis of relatedness (Fig. 3; Supplementary Fig. S2) was consistent with the polygynous mating system. This cryptic intra-population structure has certainly played a role in driving the significant FIS value and the low mean-relatedness recorded in Montes Forest.
The low migration rate and low Ne estimates suggest negligible levels of gene flow among the three sub-populations. However, both the PCA and the relatedness analysis identified an individual from Mount Tonneri showing admixed ancestry with Montes Forest. As this individual presented the mitochondrial haplotype Hpt-4 that we detected exclusively in the Mount Tonneri sub-population, we infer the individual to be a crossbred of a Montes Forest ram migrant and a Mount Tonneri ewe. This evidence suggests that a weak connectivity seems to persist, despite the very low levels of migration rate observed and the presence of artificial barriers limiting individuals’ dispersion (Fig. 1). Conversely, Mount Tonneri and Mount Lerno sub-populations showed the lowest migration rate, the highest microsatellite-based FST value, and the presence of private mtDNA haplotypes, suggesting a high degree of isolation.
The low allelic richness found in Mount Lerno and Mount Tonneri sub-populations was mirrored by a very low effective population size. Founder effect and genetic drift due to population contraction are known to cause loss of alleles and decrease of genetic variability45,46. However, recent bottleneck events are more sensitive to loss of alleles than to heterozygosity since the number of alleles drops more rapidly than allele frequencies47,48. Yet, a bottleneck event was only observed in Mount Lerno and not in Mount Tonneri. Nonetheless, the bottleneck detection procedure we implemented is known to have low prediction power when the effective size of the reduced population is too small and a new mutation-drift equilibrium is established49. We argue that the observed population contractions might be due to the devastating effects of forest fires that occurred in the past 20 years in the Mount Tonneri area (http.//www.regione.sardegna.it).
The low genetic variability currently observed in the Sardinian mouflon population might originate from the fragmentation of an original gene pool followed by long-term isolation. Originally, mouflon was widespread across Sardinia (Fig. 1) but human activities such as intensive agriculture, hunting, and high-density traffic roads, gradually lead to habitat erosion, dramatically reducing size and distribution of the population. The few areas that were only marginally affected are located in the Central-Eastern part of Sardinia, including Montes Forest and Mount Tonneri, due to their harsh orography. The genetic differentiation between Montes Forest and Mount Tonneri sub-populations could be the consequence of the landscape alterations (mainly due to a road expansion campaign started ~ 40 years ago), which may have led to the genetic depletion of the original gene pool by hindering gene exchange, and finally determining their genetic isolation. Similar situations were reported for the Californian desert bighorn sheep50 and other Sardinian macrofauna species such as wild boar and hare51,52.
Conclusions
We investigated the diversity and population structure of three Sardinian mouflon sub-populations. Particularly, we highlight the genetic distinctiveness and degree of isolation of the Montes Forest and Mount Tonneri sub-populations. We argue that these sub-populations should be considered as separate management units and preserved from contacts with genetically uncharacterized and known mouflon x sheep hybrid populations, such as the Mount Lerno population. Hybridization of wild populations with their domestic counterparts can lead to the loss of wildtype genetic integrity, outbreeding depression, and loss of adaptive features53,54,55. Morphological features are rarely informative to discriminate hybrids from pure mouflons especially when several events of backcrosses occurred55. Conversely, molecular investigations have the power to identify mixed ancestry components even with reduced panels of selected ancestry informative markers and are the only diagnostic tools able to effectively identify hybrids before carrying out restocking actions26,55. Our study prompts for initiatives to restore the habitat connectivity between Montes Forest and Mount Tonneri and promotes controlled inter-population exchange in order to improve the overall genetic diversity and counteract the effects of population fragmentation56,57. The connectivity disruption effect has been more severe in the Mount Tonneri sub-population as evidenced by the reduction in population size, Ne and allelic richness, probably due to forest fires. Actions as the implementation of biological corridors such as overpasses would allow continuous exchange of individuals among sub-populations, thus reducing the local extinction rate58. Higher gene flow leads to higher heterozygosity, which is often associated with increased fitness (heterozygote advantage), while the risk of outbreeding depression is overall minimal when the populations involved inhabit the same habitat59. This is the case for Montes Forest and Mount Tonneri sub-populations that originated in the last century from the same original gene pool, are quite close to each other and host identical habitats. The present study represents a first important step towards a deeper understanding of the genetic peculiarities of the native mouflon populations living in Sardinia. Our results highlight the deleterious effects on genetic diversity of habitat changes and fragmentation of the original gene pool. Given the key role of the Sardinian mouflon in the recolonization of continental Europe, such a knowledge is an indispensable prerequisite for the protection of its genetic identity, jeopardised by uncontrolled interaction with domestic sheep breeds.
Methods
All the animal procedures were performed in compliance with the ARRIVE guidelines and in strict accordance with the guidelines of the Ethics Committee of Sassari University, Italy, which also approved this study.
Sample collection
We collected either peripheral blood (captured mouflon) or muscular tissue samples (dead individuals) from 54 Sardinian mouflon from three distinct sites of the island during monitoring actions carried out from 2010 to 2016. A total of 39 samples (18 from Montes Forest, Orgosolo, and 21 from Mount Tonneri, Seui) were collected in the historical mouflon range. Other 15 samples were sampled from Mount Lerno (Pattada) site, where mouflons were reintroduced 30 years ago by three subsequent transfers, sourcing from Mount Albo and passing through Capo Figari and the Asinara Island (Fig. 1). Unfortunately, the collection of specimens from Mount Albo (Lula), the third historical site left on the island, was not successful due to the small population density in that area. Currently, these three sampling areas are partially isolated by recently built roads and dikes (Fig. 1) and no chance of exchange among populations exists.
DNA extraction, amplification and sequencing
Genomic DNA was extracted from blood and muscle using the GenElute Blood Genomic DNA kit (Sigma-Aldrich, Darmstadt, Germany) and the NucleoSpin Tissue XS Kit (Macherey-Nagel), respectively. Procedures were carried out according to the manufacturer's protocols. Sample quality and DNA concentration were determined via spectrophotometry using a ND-8000 (NanoDrop Technologies, Thermo Fisher Scientific Inc., Wilmington, DE). To ensure the sequence accuracy, DNA was sequenced from PCR replicates. DNA samples were used to assay 16 microsatellite loci length and the mitochondrial D-loop region polymorphisms in order to genetically characterize each group and to clarify the genetic relationships among them.
Mitochondrial DNA
The primer pair CR1-CR2 (Sanna et al., 2015) was used to amplify the mtDNA D-loop fragment. A standard 50 μL PCR mixture was used, including 200 ng DNA template, 2.5 mM MgCl2, 0.2 0 mM each dNTP, 0.20 μM each primer, 0.02 mM BSA, 1 × PCR buffer and 2 units Taq DNA Polymerase (Sigma-Aldrich), according to Mereu et al.32. PCR amplifications were performed in a Gradient Thermocycler (Eppendorf) by an initial denaturation of 95 °C for 3 min, followed by 30 cycles of 95 °C for 50 s, 60 °C for 30 s, and 72 °C for 1 min.
PCR products were sequenced using the same primers on an ABI 3130 Genetic Analyzer (Applied Biosystem). Sequencing reactions were carried out following the manufacturer's recommendations (BigDye Terminator 3.1 Cycle Sequencing Kit—Applied Biosystem) and purified through the SigmaSpin Post—Reaction Clean—UP Columns (Sigma-Aldrich).
Raw sequencing data were processed by means of the KB base-calling algorithm implemented in the Sequencing Analysis Software 5.3.1 (Applied Biosystem).
Haplotypes and population structure analysis
Sequence alignment and data formatting, including trimming to a core length of 439 bp and resolving the ambiguities (N) in base-calling, were performed using BioEdit60.
The amount of genetic variation among the three sub-populations, including the number of polymorphic sites (S) and haplotypes (h), the haplotype diversity (Hd) and nucleotide diversity (π), were estimated using DnaSP 6.10.0361.
The analysis of molecular variance (AMOVA) and the calculation of the coefficient of differentiation (FST) were carried out using Arlequin 3.562.
Genetic relationships among haplotypes were investigated by a median joining network (MJN) on the nucleotide sequence matrix of 91 D-loop sequences. To explore the evolutionary relationships between the Sardinian mouflon and other O. gmelini species, the dataset was combined with a total of 34 homologous sequences of mouflons from Cyprus (n = 3), Corsica (n = 2), mainland Europe (n = 3), Anatolia (n = 16) and Asia (n = 10). These sequences were a representative sub-sample of all the haplotypes deposited in GenBank and were selected, one for each haplotype, on the basis of similarity detected at the BLAST analysis. Three O. vignei sequences were also included as outgroup (Supplementary Table S5). The MJN was constructed in PopART 1.750 [http://popart.otago.ac.nz].
The pairwise genetic distances were estimated with the software MEGA 7.0.1463 using 10,000 bootstrap replicates. The Tamura-Nei (TN93)64 model of nucleotide substitution with uniform evolutionary rates among sites was chosen to estimate genetic distance between haplotypes on the basis of the lower Bayesian information criterion (BIC) score.
Microsatellites
We genotyped 54 samples at 14 microsatellites designed for domestic sheep (O. aries) and cattle (Bos taurus) obtained from public databases (NCBI genome database, http://www.ncbi.nlm.nih.gov; ARKdb database, http://www.thearkdb.org). Markers were amplified by PCR in 25 µl reaction mix containing: 10 μl of DNA (1 ng/μl) as a template, 1X PCR Buffer, 1.5 mM of each dNTP, 0.5 μM of each primer, 1 unit of Taq Polymerase and 1.5 mM MgCl2. PCR amplifications were performed in a Gradient Thermocycler (Eppendorf) by an initial denaturation of 95 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 48–58 °C for 1 min (Supplementary Table S6) and 72 °C for 1 min, followed by a final step of 72 °C for 5 min.
Microsatellite products were analysed on an ABI PRISM 3100 DNA Analyzer (Applied Biosystems) and data processed using GeneScan 3.1 and Genotyper 2.565. Identification of possible genotyping errors due to null alleles, short allele dominance and typographic errors, were tested using Micro-Checker software66 (http://www.microchecker.hull.ac.uk). Allelic richness and FIS values were calculated using Fstat 2.9.367. Differences between populations in allelic richness were tested using the Fisher exact test method, after assessing for normality of the data and variance homogeneity (Minitab 17 statistical software, Minitab, Inc. State College, PA), while FIS significance was tested by 95% confidence interval with 1000 bootstrap replicates by Fstat 2.9.3. Allele frequency, gene diversity (expected heterozygosity—He, Nei 1973), observed heterozygosity (Ho) and Hardy–Weinberg equilibrium (HWE) deviation analyses by population and locus were calculated using the Fisher exact test68 in a contingency table of arbitrary size, as implemented in Arlequin 3.562. The exact P-values were obtained using a Markov Chain Monte Carlo (MCMC) simulation with 10,000 dememorization steps, 500 batches, and 5000 iterations.
Genetic differentiation (FST) used as inter-population indices to assess the relationships among the three mouflons groups was calculated by permuting the genotypes or individuals between the sub-populations. The significance of the genetic distances was tested by Weir & Cockerham69 as implemented in Arlequin 3.5 (10,000 permutations).
Population genetic structure was investigated using the analysis of molecular variance (AMOVA) by Arlequin 3.5.
A principal component analysis (PCA) was performed to investigate the ordinal relationships between populations and individuals, using the ade4 1.7-16 package70 of the R software71.
Recent migration rates among sub-populations were assessed using a Bayesian Markov Chain Monte Carlo (MCMC) analysis implemented in BayesAss 1.372 which allows estimation of the rate and direction of recent dispersal. The MCMC method was run for 10,000,000 iterations. Delta values (i.e., maximum parameter change per iteration) were left as default73.
Pairwise relatedness (r) between individuals within populations based on the method of Lynch and Ritland74 with the 2x option was estimated using GenAlEx 6.50175 to detect related individuals within the same sub-population. This method assumes values from − 1 (completely unrelated individuals) to 1 (identical twins). To calculate pairwise relatedness, we used the allele frequency of the total population of assembled genotypes with 9999 permutations and r frequencies were plotted to evaluate its distribution. Distribution factors such as median and mean were compared by χ2 test and ANOVA test respectively, using Minitab 17. Relatedness estimates higher than 0.5 observed in each population were compared in the whole sample by means of Pearson χ2 test (Minitab 17). To assess the presence of sub-structures within sub-populations a matrix of pairwise relatedness distances between individuals was computed by Genalex 6.501 and a multivariate analysis was performed using the Cluster Variables function implemented in Minitab 17. The results were graphically expressed by a UPGMA tree (Minitab 17).
Bottleneck events were detected from allele frequency data by the BOTTLENECK software76 assuming that microsatellite mutation followed the Infinite Allele Model (IAM), Stepwise Mutation Model (SMM) or Two-Phase Model (TPM)77. In TPM we used 95% single-step mutational events at 12% variance. To determine the significance in the heterozygosity excess we performed the one-tailed Wilcoxon test. A mode-shift test was carried out to detect a distortion on the expected L-shaped distribution of allele frequency and to determine whether the population is under mutation drift equilibrium (shaped curve) or not (mode shift)78.
We estimated the effective population size using the Linkage disequilibrium method implemented in NeEstimator 279. This method has been proven relatively robust in assessing the effective population size (Ne) using microsatellite data80. As rare alleles could result in biased Ne, we calculated three estimates for Ne by excluding allele frequencies of less than (Pcrit) 0.05, 0.02, or 0.01. The 95% CI was estimated by the parametric method81.
References
Hiendleder, S., Kaupe, B., Wassmuth, R. & Janke, A. Molecular analysis of wild and domestic sheep questions current nomenclature and provides evidence for domestication from two different subspecies. Proc. R. Soc. B Biol. Sci. 269, 893–904 (2002).
Meadows, J. R. S., Cemal, I., Karaca, O., Gootwine, E. & Kijas, J. W. Five ovine mitochondrial lineages identified from sheep breeds of the near east. Genetics 175, 1371–1379 (2007).
Zeder, M. A. Domestication and early agricolture in the Mediterranean Basin: origins, diffusion, and impact. PNAS 105, 11597–11604 (2008).
Chessa, B. et al. Revealing the history of sheep domestication using retrovirus integrations. Science (80-) 324, 532–536 (2009).
Sanna, D. et al. The first mitogenome of the Cyprus mouflon (Ovis gmelini ophion): New insights into the phylogeny of the genus Ovis. PLoS ONE 10, e0144257 (2015).
Ciani, E. et al. On the origin of European sheep as revealed by the diversity of the Balkan breeds and by optimizing population-genetic analysis tools. Genet. Sel. Evol. 52, 1–14 (2020).
Barbato, M. et al. Genomic signatures of adaptive introgression from European mouflon into domestic sheep. Sci. Rep. 7, e7623 (2017).
Mereu, P. et al. Identification of an ancestral haplotype in the mitochondrial phylogeny of the ovine haplogroup B. PeerJ 2019, e7895 (2019).
Apollonio, M., Luccarini, S., Giustini, D., Scandura, M. & Ghiandai, F. Carta delle vocazioni faunistiche della Sardegna. http://www.sardegnaambiente.it/documenti/18_269_20121204133805.pdf (2005).
Zimmerman, M. MOUFFLON - Preservation and spread of the Corsican moufflon populations within Corsica - LIFE03 NAT/F/000099. https://ec.europa.eu/environment/life/project/Projects/index.cfm?fuseaction=search.dspPage&n_proj_id=2500&docType=pdf (2008).
Fleba, L., Serra, R. & Manca, M. Censimento del Muflone (Ovis aries Linnaeus 1758) nei territori gestiti dall’Ente Foreste della Sardegna. https://www.sardegnaforeste.it/sites/default/files/Censimento%20Muflone_ente%20Foreste_2015.pdf (2015).
Poplin, F. Origine nouvelle perspective paléontologique par marronnage. Ann. Genet. Sel. Anim. 11, 133–143 (1979).
Levine, M. La fauna di Filiestru. in La grotta di Filiestru a Bonu Ighinu, Mara (SS). (ed. Dessì) (1983).
Ciuti, S., Pipia, A., Grignolio, S., Ghiandai, F. & Apollonio, M. Space use, habitat selection and activity patterns of female sardinian mouflon (Ovis orientalis musimon) during the lambing season. Eur. J. Wildl. Res. 55, 589–595 (2009).
Ruiz-Fons, F., Sánchez-Matamoros, A., Gortázar, C. & Sánchez-Vizcaíno, J. M. The role of wildlife in bluetongue virus maintenance in Europe: Lessons learned after the natural infection in Spain. Virus Res. 182, 50–58 (2014).
Beccu, E. La conservazione e la gestione della fauna selvatica nelle foreste demaniali della regione sarda. in Atti 1° Convegno regionale sulla fauna selvatica in Sardegna. (eds. Cossu, S., Onida, P. & Torre, A.) 187–200 (Edizioni del Sole e Amministrazione Provinciale di Oristano, 1993).
Franklin, I. Evolutionary change in small populations. In Conservation Biology: An Evolutionary–Ecological Perspective (eds. Soule’, M. E. & Wilcox, B. A.) 135–150 (Sinauer Associates, 1980).
Brooks, S. P., Catchpole, E. A. & Morgan, B. J. T. Bayesian animal survival estimation. Stat. Sci. 15, 357–376 (2000).
van der Valk, T., de Manuel, M., Marques-Bonet, T. & Guschanski, K. Estimates of genetic load in small populations suggest extensive purging of deleterious alleles. bioRxiv (2019) https://doi.org/10.1101/696831.
Hailer, F. et al. Bottlenecked but long-lived:High genetic diversityretained in white-tailedeagles upon recoveryfrom population decline. Biol. Lett. 2, 316–319 (2006).
Kaeuffer, R., Coltman, D. W., Chapuis, J. L., Pontier, D. & Réale, D. Unexpected heterozygosity in an island mouflon population founded by a single pair of individuals. Proc. R. Soc. B Biol. Sci. 274, 527–533 (2007).
Portanier, E. et al. Introduction history overrides social factors in explaining genetic structure of females in Mediterranean mouflon. Ecol. Evol. 7, 9580–9591 (2017).
Pliny The Elder. Natural History. (Betascript Publishing, 2010).
Uloth, W. To the history of the distribution, introduction and cross-breeding of the Tyrrhenis mouflon in Europe and oversea. Acta Theriol. (Warsz) 17, 412–413 (1972).
Weller, K. The status of mouflon (Ovis musimon) in Europe. In Proceedings of the Third International Symposium on Mouflon (eds. A, N. & W, U.) 114–140 (András Náhlik, 2001).
Schröder, O. et al. Limited hybridization between domestic sheep and the European mouflon in Western Germany. Eur. J. Wildl. Res. 62, 307–314 (2016).
Lorenzini, R., Cabras, P., Fanelli, R. & Carboni, G. L. Wildlife molecular forensics: Identification of the Sardinian mouflon using STR profiling and the Bayesian assignment test. Forensic Sci. Int. Genet. 5, 345–349 (2011).
Meadows, J. R. S., Hiendleder, S. & Kijas, J. W. Haplogroup relationships between domestic and wild sheep resolved using a mitogenome panel. Heredity (Edinb) 106, 700–706 (2011).
Hiendleder, S., Lewalski, H., Wassmuth, R. & Janke, A. The complete mitochondrial DNA sequence of the domestic sheep (Ovis aries) and comparison with the other major ovine haplotype. J. Mol. Evol. 47, 441–448 (1998).
Lancioni, H. et al. Phylogenetic relationships of three Italian merino-derived sheep breeds evaluated through a complete mitogenome analysis. PLoS ONE 8, 1–10 (2013).
Morelli, L. et al. Mitochondrial DNA lineages of Italian Giara and Sarcidano horses. Genet. Mol. Res. 13, 8241–8257 (2014).
Mereu, P. et al. The complete mtDNA sequence of the griffon vulture (Gyps fulvus): Phylogenetic analysis and haplotype frequency variations after restocking in the Sardinian population. Biol. Conserv. 214, 195–205 (2017).
Gonzalez-Suarez, M., McCluney, K. E., Aurioles, D. & Gerber, L. R. Incorporating uncertainty in spatial structure for viability predictions: A case study of California sea lions (Zalophus californianus californianus). Anim. Conserv. 9, 219–227 (2006).
Portanier, E. et al. Landscape genetics matches with behavioral ecology and brings new insight on the functional connectivity in Mediterranean mouflon. Landsc. Ecol. 33, 1069–1085 (2018).
Strnad, M. et al. SUB-TASK IX. Migration corridors. https://conservationcorridor.org/cpb/Strnad_et_al_2013.pdf (2013).
Cugnasse, J. M. Révision taxinomique des mouflons des îles méditerranéennes. Mammalia 58, 507–512 (1994).
Demirci, S. et al. Mitochondrial DNA diversity of modern, ancient and wild sheep (Ovis gmelinii anatolica) from Turkey: New insights on the evolutionary history of sheep. PLoS ONE 8, e81952 (2013).
Kalinowski, S. T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 5, 539–543 (2004).
Pruett, C. L. & Winker, K. The effects of sample size on population genetic diversity estimates in song sparrows Melospiza melodia. J. Avian Biol. 39, 252–256 (2008).
Hale, M. L., Burg, T. M. & Steeves, T. E. Sampling for microsatellite-based population genetic studies: 25 to 30 individuals per population is enough to accurately estimate allele frequencies. PLoS ONE https://doi.org/10.1371/journal.pone.0045170 (2012).
Geist, V. Mountain Sheep: A Study in Behavior and Evolution. (University of Chicago Press, 1971).
Greenwood, P. J. Mating systems, philopatry and dispersal in birds and mammals. Anim. Behav. 28, 1140–1162 (1980).
Keller, L. & Waller, D. Inbreeding effects in wild populations. Trends Ecol. Evol. 17, 230–241 (2002).
Kruuk, L. E. B., Sheldon, B. C. & Merilä, J. Severe inbreeding depression in collared flycatchers (Ficedula albicollis). Proc. R. Soc. B Biol. Sci. 269, 1581–1589 (2002).
Dlugosch, K. M. & Parker, I. M. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449 (2007).
Nei, M., Maruyama, A. T. & Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution (N. Y) 29, 1–10 (1975).
Greenbaum, G., Templeton, A. R., Zarmi, Y. & Bar-David, S. Allelic richness following population founding events—A stochastic modeling framework incorporating gene flow and genetic drift. PLoS ONE 9, 1–23 (2014).
Comps, B., Gömöry, D., Letouzey, J., Thiébaut, B. & Petit, R. J. Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157, 389–397 (2001).
Larsson, J. K., Jansman, H. A. H., Segelbacher, G., Höglund, J. & Koelewijn, H. P. Genetic impoverishment of the last black grouse (Tetrao tetrix) population in the Netherlands: Detectable only with a reference from the past. Mol. Ecol. 17, 1897–1904 (2008).
Epps, C. W. et al. Highways block gene flow and cause a rapid decline in genetic diversity of desert bighorn sheep. Ecol. Lett. 8, 1029–1038 (2005).
Scandura, M., Iacolina, L., Cossu, A. & Apollonio, M. Effects of human perturbation on the genetic make-up of an island population: The case of the Sardinian wild boar. Heredity (Edinb). 106, 1012–1020 (2011).
Canu, A. et al. Differentiation under isolation and genetic structure of Sardinian hares as revealed by craniometric analysis, mitochondrial DNA and microsatellites. J. Zool. Syst. Evol. Res. 50, 328–337 (2012).
Dovc, P. et al. Prospects and challenges for the conservation of farm animal genomic resources, 2015–2025. Front. Genet. 6, 314 (2015).
Iacolina, L. & Safner, T. Hybridisation in European ungulates: an overview of the current status, causes, and consequences. Mamm. Rev. 49, 45–59 (2019).
Somenzi, E., Ajmone-Marsan, P. & Barbato, M. Identification of ancestry informative marker (AIM) panels to assess hybridisation between feral and domestic sheep. Animals 10, 582 (2020).
Wiens, J. A., Stenseth, N. C., Van Horne, B. & Ims, R. A. Ecological mechanisms and landscape ecology. Oikos 66, 369–380 (1993).
Lindenmayer, D. et al. A checklist for ecological management of landscapes for conservation. Ecol. Lett. 11, 78–91 (2008).
Rosenberg, D. K., Noon, B. R. & Meslow, E. C. Biological corridors: Form, function, and efficacy. Linear conservation areas may function as biological corridors, but they may not mitigate against additional habitat loss. Bioscience 47, 677–687 (1997).
Miller, J. M., Poissant, J., Hogg, J. T. & Coltman, D. W. Genomic consequences of genetic rescue in an insular population of bighorn sheep (Ovis canadensis). Mol. Ecol. 21, 1583–1596 (2012).
Hall, T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526 (1993).
Abbas, M. et al. Genetic polymorphisms of TLR4 and MICA are associated with severity of Trachoma Disease in Tanzania. Autoimmune Infect. Dis. https://doi.org/10.16966/2470-1025.116 (2016).
Van Oosterhout, C., Weetman, D. & Hutchinson, W. F. Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol. Ecol. Notes 6, 255–256 (2006).
Goudet, J. Fstat (Version 1.2): A computer program to calculate F-statistics. J. Hered. 86, 485–486 (1995).
Guol, S. W. & Thompson, E. A. Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48, 361–372 (1992).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution (N. Y) 38, 1358–1370 (1984).
Dray, S. & Dufour, A. B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20 (2007).
R Core Team. R: A language and environment for statistical computing. (2010).
Wilson, G. A. & Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163, 1177–1191 (2003).
Beerli, P. & Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. U. S. A. 98, 4563–4568 (2001).
Lynch, M. & Ritland, K. Estimation of pairwise relatedness with molecular markers. Genetics 152, 1753–1766 (1999).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539 (2012).
Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014 (1996).
Piry, S., Luikart, G. & Cornuet, J. M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503 (1999).
Luikart, G., Allendorf, F. W., Cornuet, J. M. & Sherwin, W. B. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered. 89, 238–247 (1998).
Do, C. et al. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14, 209–214 (2014).
Waples, R. S. & Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 3, 244–262 (2010).
Jones, A. T., Ovenden, J. R. & Wang, Y. G. Improved confidence intervals for the linkage disequilibrium method for estimating effective population size. Heredity (Edinb) 117, 217–223 (2016).
Funding
This work was supported by Fondazione Banco di Sardegna and “Fondo di Ateneo per la ricerca 2019”, University of Sassari.
Author information
Authors and Affiliations
Contributions
V.S, P.M., M.B., M.P., L.M., S.N. and G.G.L. made contributions to the conception and design of the work; G.B. prepared samples and collected data; V.S. and P.M. performed all experiments; V.S., P.M., M.B., M.P. and G.G.L. performed analyses and data interpretation; V.S., P.M., M.P. and G.G.L wrote the original draft; V.S., P.M., M.B., M.P., L.M., S.N. and G.G.L. revised the manuscript; L.M. S.N. and G.G.L. secured funding; V.S., P.M., M.P. and G.G.L. supervised the project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Satta, V., Mereu, P., Barbato, M. et al. Genetic characterization and implications for conservation of the last autochthonous Mouflon population in Europe. Sci Rep 11, 14729 (2021). https://doi.org/10.1038/s41598-021-94134-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94134-3
This article is cited by
-
Phenotype transition from wild mouflon to domestic sheep
Genetics Selection Evolution (2024)
-
Detection of selective sweep in European wild sheep breeds
3 Biotech (2024)
-
Genetic variation of European mouflon depends on admixture of introduced individuals
Mammal Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
