
Abstract
A method is described for the detection of certain nucleotide modifications adjacent to the 5’ 7-methyl guanosine cap of mRNAs from individual genes. The method quantitatively measures the relative abundance of 2’-O-methyl and N6,2’-O-dimethyladenosine, two of the most common modifications. In order to identify and quantitatify the amounts of N6,2’-O-dimethyladenosine, a novel method for the synthesis of modified adenosine phosphoramidites was developed. This method is a one step synthesis and the product can directly be used for the production of N6,2’-O-dimethyladenosine containing RNA oligonucleotides. The nature of the cap-adjacent nucleotides were shown to be characteristic for mRNAs from individual genes transcribed in liver and testis.
Similar content being viewed by others

Introduction
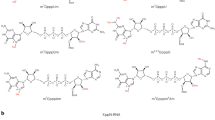
In most eukaryotes, polymerase II (Pol II) dependent transcripts are modified co-transcriptionally at their 5’end by the addition of a 7-methyl guanosine (m7G) cap to the first nucleoside of the nascent transcript. If no additional modifications are made to the cap-adjacent nucleotides, the structure is referred to as a cap0. In yeast and plants, only cap0 structures are found, however, in animals, modifications of the two nucleotides adjacent to the m7G are possible The methylation of the first nucleotide on the ribose residue (Fig. 1a) will form a cap1 structure1. Cap1 messages can be converted to cap2 structures if a further 2’ribose methylation takes place on the next nucleotide following the cap1 (Fig. 1b). These methylation steps are sequential and carried out by nuclear located methylases1,2,3. In a transcript where the first nucleotide is an adenosine in a cap1 structure, a further methylation of the 2’-O-dimethyladenosine (Am) at the N6 position of the adenine can take place to give N6,2’-O-dimethyladenosine (m6Am) (Fig. 1c)4. The methyltransferase that carries out this modification has been partially characterized and appears to be predominantly located in the cytoplasm5. Transcription start sites of focused promoters are usually contained within an initiator motif (Inr) of general sequence YYANWYY (Y = pyrimidine, N-any nucleotide, W = A or T) where the A is the principal start site and neighbouring nucleotides may be used to varying degrees6. Since, A is often the first nucleotide after the m7G cap, its modification may have a functional role. In order to study the effect of m6Am present in cap1 messages, the chemical synthesis of RNA oligonucleotide sequences that contain m6Am in well-defined positions, is necessary. A number of methods exist in the literature for the preparation of N6-methylated adenosine derivatives, with the Dimroth rearrangement being perhaps the most well known7,8,9. This transformation relies upon an initial N1-methylation of the adenine ring followed by an alkali-mediated rearrangement to give the N6-methylated adenine product. An alternative method for accessing N6-alkylated adenines has been developed, which involves the nucleophilic aromatic substitution of adenine derivitives, that are activated at the 6-position, with amine nucleophiles (i.e. MeNH2)10,11,12,13. In order to obtain the N6-methylated phosphoramidite reagents from these adenine derivatives, a number of additional synthetic steps are required, which results in a 6 to 8 step synthesis being needed to produce each phosphoramidite reagent10,11,12,13. Such approaches are not realistic other than in synthetic chemistry laboratories.

Cap adjacent modification in higher Eukaryotes.
Formation of the cap1 structure takes place by 2’-O –methylation on the ribose residue on the first nucleotide adjacent to the m7G cap. The example shows a structure with adenosine as the first nucleotide, adjacent to the m7G cap (a). The cap1 structure can then be converted to a cap2 by a further 2’-O-ribose methylation on the following nucleotide (b), or converted to a cap1 m6Am by placing a further methyl group on the adenosine in the N6 position (c). The figure is a schematic representation of the mRNA cap structures. The yellow structure represents 7-methyl-guanine, the green represents adenine and the grey pentagon is ribose. On the ribose only the methylated functional groups are shown.
An interaction of m7G capped transcripts with the nuclear cap-binding complex (CBC) is important for promoting correct splicing14 and 3’end formation15. After export from the nucleus, translation of most mRNAs is initiated with the recognition of the m7G cap by eukaryotic translation initiation factor 4E (eIF4E)16. This cap binding step is usually rate-limiting for translation. The m7G cap also protects the message from 5’ to 3’ exonuclease digestion. Compared to the well characterised role of m7G, relatively little is known regarding the function of 2’ ribose methylation on the following nucleotides. The presence of a cap1 structure may promote binding to ribosomes17 and can increase translational efficiency in vitro18,19. Many animal viruses encode their own cap1 methylases20,21 and this may also help them to limit host immune responses22,23. Whilst viral cap1 methylases are common viral cap2 methylases and m6Am methylases have not been reported. However, m6Am and cap2 structures can be found in viral mRNAs in vivo, presumably as a result of the action of the host cell’s enzymes24. The functional consequences for an mRNA of possessing m6Am in a cap1 or a cap2 structure are not known.
In order to investigate the roles of these cap-associated modifications, we have developed enzymatic techniques, in combination with thin layer chromatography (TLC) that allow the detection of m6Am and other cap nucleotides. We also describe a direct synthetic method for the preparation of the desired m6Am phosphoramidite reagent in a single step from commercially available starting material.
Through the use of this synthetic substrate we have demonstrated that a thin layer chromatography (TLC) method is quantitative for determining m6Am:Am ratios and allows the relative proportions of Am and m6Am to be rapidly determined for mRNAs from different cell types and also for caps of messages from individual genes. We further give evidence that the relative proportion of m6Am varies in a characteristic way for mRNA from different mouse organs.
Results
Synthesis and detection of N6, 2’-O- dimethyladenosine
We have previously used TLC assays to detect and quantify N6-methyladenosine (m6A)25, a modification found internally in the mRNA of many eukaryotes26,27,28,29. This method was modified to label and detect only the first nucleotide after the cap. In order to use this approach to compare the relative proportions of m6Am and Am in cap structures from different sources, it was first necessary to establish the mobility of m6Am in the TLC assay and to demonstrate that T4 polynucleotide kinase does not preferentially label either of these nucleotides. As neither m6Am triphosphate or m6Am phosphoramidite are commercially available, a novel method for its synthesis and incorporation into RNA oligonucleotides was developed.
In order to get quick access to the required m6Am phosphoramidite building block for solid-phase oligoribonucleotide synthesis, we performed a selective one-step methylation of the commercially available 2’-OMe-Bz-A-CE phosphoramidite. Due to its high reactivity and sensitivity towards acids and bases, Aritomo’s relatively mild phase transfer catalysis (PTC) method was used30. Under these conditions (iodomethane, sodium hydroxide, tetrabutylammonium bromide) the N6-methylated product was obtained as the major product in 56% yield, with the N1-methylated isomer being formed as a byproduct in 25% yield. Due to their different Rf values, the N6- and N1-methylated products were easily separated via column chromatography and the purified m6Am phosphoramidite was used directly for RNA synthesis.
All oligoribonucleotides were synthesized using the standard protocol for solid-phase RNA synthesis with a 15 min coupling time per nucleotide. The last coupling step was followed by a standard DMTr-off procedure, the oligoribonucleotides were removed from the solid support, deprotected, precipitated and desalted. MALDI mass spectrometry was used to confirm the mass of all RNA oligonucleotides produced
Thin layer chromatography of modified adenosines
Radioactively 5’ end labelled oligonucleotides SK-526 (m6AmGGGCUGCU) and SK-524 (AmGGGCUGCU) were digested to release pm6Am and pAm then mixed with a combination of 5’ radioactive labelled pm6A and unmodified nucleotide monophosphates for which relative mobilities are well characterised. This mixture was separated by 2D TLC using the solvent systems previously described25 to establish the position of pm6Am (Fig. 2).

Relative mobility of modified adenosines using 2 dimensional thin layer chromatography.
RNA oligonucleotide fragments of known compositions were labelled at their 5’ ends with32P, digested to 5’ monophosphates and separated by 2 dimensional thin layer chromatography in order to establish the mobility of pm6Am relative to other nucleotides. Adenosine and its methylated derivatives are indicated by shading.
To show that T4 polynucleotide kinase does not preferentially label m6Am or Am, the RNA oligonucleotides SK-524 and SK-526 were mixed in different ratios, end labelled, digested with P1 nuclease and separated by TLC. The spots corresponding to pm6Am and pAm were then quantified using phosphorimaging. The results demonstrated that both nucleotides are labelled with equal efficiency by T4 polynucleotide kinase (Fig. 3)

T4 polynucleotide kinase does not preferentially label m 6 Am or Am containing ends.
The oligonucleotides SK-526 (m6Am) and SK-524 (Am) were mixed in ratios of 5:1, 2:1, 1:1. 1:2 and 1:5, end labelled using T4 polynucleotide kinase, digested to 5’ phosphate mononucleotides and separated by TLC. The relative intensities of the resulting spots were quantified using phosphorimaging. The calculated percentage of m6Am closely matched the actual m6Am percentage.
Labelling and analysis of the first cap adjecent nucleotide
To label the first nucleotide following the m7G, poly(A) RNA was prepared from various mouse organs then digested with tobacco acid pyrophosphatase to remove the m7G cap. The exposed 5’ ends were dephosphorylated with alkaline phosphatase and after phenol/chloroform extraction and ethanol precipitation, the mRNA transcripts were radiolabelled at their 5’ end using T4 polynucleotide kinase in the presence of >20 fold excess of [γ-32P] ATP. The labelled RNA was digested to monophospho-nucleotides by P1 nuclease prior to TLC separation. Using this method, new spots corresponding to the 2’-O methylated nucleotides are apparent in the labelled samples after cap removal (Fig 4A). A spot corresponding to pm6Am is readily detectable in all mRNA samples tested and with pm6Am:pAm ratios of between 15:1 (brain) and 2:1 (liver) it appears that m6Am is more prevalent at the cap1 than is Am (Fig. 4A). Under these labelling conditions, where both ATP and polynucleotide kinase are in excess, the intensity of the spots corresponding to the unmodified nucleotides, as well as the other 2’ methylated nucleotides, is indicative of their actual relative abundance in the de-capped mRNA sample (Supplementary Figure S1 online).

Relative abundance of modified adenosines in mRNA from different mouse organs and in individual mRNAs.
(A) 2’-O-methylated nucleotides are present in the mRNA populations from various mouse organs. Arrow indicates m6Am. Different organs have characteristic m6Am:Am ratios; brain (15:1), kidney (2.4:1), liver (2:1), testis (10:1). (B) Transcripts from individual genes have characteristic nucleotides or nucleotide modifications at the first nucleotide position; Pabpc1 (poly(A) binding protein cytoplasamic 1), Apoa1 (apolipoprotein A-I), Prm2, (protamine 2), Alb (albumin). (C) Schematic indicating the relative position of the nucleotide spots (left) and the location of the cold single stranded target sequences on the hybridisation template (right). Single stranded DNA from the yeast gene IME2 was used as a negative control DNA in the hybridisations.
Analysis of cap structures for mRNA transcripts from individual genes
In order to assay modifications on the first transcribed nucleotide for messages from individual genes, mRNA from liver and testis was de-capped and end-labelled as described above then fragmented to ∼120 nt. This was then hybridised to single stranded DNA targets corresponding to the 5’ region of selected messages. These DNA targets were first cross-linked to 2 mm × 2 mm teeth cut from a Hybond N+ membrane. After hybridization and washing, the membrane was subjected to phosphorimaging (Fig. 4B). The individual teeth containing the labelled mRNA gene-specific fragments were then removed and digested to nucleotide 5’ monophosphates using P1 nuclease. These samples were individually spotted onto TLC plates and developed as described (Fig. 4B). Four mRNAs were chosen for analysis, apolipoprotein A-I (Apoa1, BC012253), albumin (Alb, BC024643), protamine 2 (Prm2, BC049612) and poly(A) binding protein cytoplasmic 1 (Pabpc1, BC046233). Alb and Apoa1 are predominantly liver expressed messages, whereas Prm2 shows testis specific expression and Pabpc1 is expressed highly in both organs. Pabpc1 may be subjected to translational regulation, but unlike Pabpc2, it is present in actively translating polyribosomes of mouse testicular cells31. For both Alb and Apoa1, the labelled nucleotide in the cap adjacent position included A, G, U and C, as well as m6Am and Am. This is consistent with multiple alternative transcription start sites at and around the Inr32. In both cases, Am and m6Am appeared as the major nucleotide modifications; unlike the mixed mRNA starting material, 2’-O-methylcytosine was not present (compare Fig. 4A, B). The m6Am:Am ratios were 1.65:1 and 1.4:1 for Apoa1 and Alb respectively, which is only slightly lower compared to that seen for the liver mRNA population as a whole. The Prm2 mRNAs from testis tissues almost exclusively had m6Am at the cap adjacent position (Fig. 4B). With an m6Am:Am ratio of 40:1, this was four times larger than the m6Am:Am ratio for the testis mRNA population as a whole. In contrast the cap adjacent nucleotides of Pabpc1 transcripts were predominantly unmodified, although a pronounced 2’-O-methyluridine was apparent in the sample from testis (Fig. 4B).
Discussion
Core promoters of Pol II transcribed genes may be focused, directing transcription initiation at a single site or cluster of adjacent sites, or they may be dispersed and have multiple start sites over a region of 50–100 nucleotides6. In case of focused promoters the transcription start sites are contained within and around the Inr (YYANWYY), where the A is the principal start site and surrounding nucleotides are utilised to varying degrees6. Recent high-throughput sequencing from full length mRNAs, suggests that purine instead of A may be a better representation of the Inr consensus. However, most core promoters have several closely arrayed transcription start sites with different initiation rates rather than initiating at a single nucleotide32. Thus, most genes will give rise to a population of mRNAs that differ in their starting nucleotides. For example the Prm2 message initiates predominantly at the central A of the Inr as well as a second A 3 nucleotides upstream, whereas, Apoa1 strongly initiates at the A of the Inr but also uses surrounding U, G, C and sites at a significant level (Supplementary Figure S2 online) and (Fig. 4B).
Conversion of an adenosine following the m7G cap to Am (cap1) can be followed by further conversion to m6Am (Fig. 1a and 1c). It is not known if this Am to m6Am conversion preferentially targets certain messages, or if the Am:m6Am ratio for messages from any one gene is merely a reflection of the general m6Am methylase activity of a particular cell type. The enzyme responsible for the formation of m6Am has been partially characterised5. It appears to be a cytosolic enzyme and is distinct from METTL3/MT-A70, the nuclear enzyme responsible for the formation of m6A at internal positions within mRNA33.
Whilst there is evidence that cap1 methylation may increase translation efficiency18,19, the consequences of converting a cap1 Am to m6Am are largely unknown. Some crystallographic observations suggest that the first nucleotide after the m7G may influence interaction with eIF4E or other cap binding proteins. Crystallographic analysis of eIF4E in a complex with the dinucleotide m7GpppG, show the G in a disordered arrangement34. However, with m7GpppA as substrate, the A interacts with a C-terminal loop of the protein35. A key interaction of this C-loop is a hydrogen bond between Thr205 and the amino group in the N6 position of the adenosine. It is this amino group that is targeted for methylation by the m6Am methylase. Thus, N6 methylation of a cap1 adenosine might influence the flexibility and orientation of the eIF4E C-terminal loop region. The ability to synthesize RNA oligonucleotides that containe m6Am in well-defined positions should facilitate future structure-function analysis. Therefore a direct synthetic method for the preparation of the desired m6Am phosphoramidite reagent, in a single step from commercially available starting material via direct N6-methylation, dramatically improves access to these materials for biological evaluation.
Methods
Preparation of 2’-OMe-Bz-m6A-CE phosphoramidite
5′-(4,4′-Dimethoxytrityl)-6-N-benzoyl-6-N-methyl-adenosine,2′-O-methyl-3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (2’-OMe-Bz-m6A-CE phosphoramidite) was prepared as follows: tetrabutylammonium bromide (260 mg, 807 μmol) and aq. NaOH (1M, 7 ml) were added to a stirring solution of 2’-OMe-Bz-A-CE phosphoramidite (Link Technologies, 708 mg, 797 μmol) and iodomethane (200 μl, 3.21 mmol) in dichloromethane (7 ml). After vigorous stirring for 30 min, ether and water were added and the resulting layers were separated. The aqueous layer was extracted with ether, the combined organic extracts were dried over Na2SO4 and the volatiles were removed in vacuo. 2’-OMe-Bz-m6A-CE phosphoramidite was readily purified by column chromatography (1:1 petrol/ether→9:1 ether/methanol) affording 2’-OMe-Bz-m6A-CE phosphoramidite (403 mg, 56%, white foam, 1:1 mixture of diastereoisomers) and 2’-OMe-Bz-m1A-CE phosphoramidite (182 mg, 25%, white foam, 1:1 mixture of diastereoisomers). Data for 2’-OMe-Bz-m6A-CE phosphoramidite and for 2’-OMe-Bz-m1A-CE phosphoramidite are presented in Supplementary Table S1. online.
Synthesis of oligoribonucleotides SK-524 and SK-526
Oligoribonucleotides SK-524(AmGGGCUGCU) and SK-526(m6AmGGGCUGCU) were synthesized on an ABI 394 DNA/RNA synthesizer and mass spectra were recorded using a Bruker Ultraflex III mass spectrometer via MALDI-TOF. Columns (SynBaseTM CPG 1000Å, RNA: 0.2 µmol), apart from the 2’-OMe-Bz-m6A-CE phosphoramidite, standard RNA-phosphoramidites and reagents for the synthesizer were purchased from Link Technologies Ltd., MeNH2 solution (33 wt.% in ethanol) was obtained from Fluka, NEt3·3HF, N-methylpyrrolidinone (NMP), 3-hydroxypicolinic acid (HPA) and DowexTM 50WX8-200 were purchased from Aldrich, illustra Nap™-10 columns were obtained from GE Healthcare Europe GmbH. Dichloromethane and acetonitrile were freshly distilled from CaH2 before use on the synthesizer.
The RNA oligonucleotides were synthesized using a standard 0.2 µM scale protocol, but with a 15 min coupling time for each nucleotide addition step. The polymer-bound oligoribonucleotide was transferred from the synthesis column to a 1.5 ml microfuge tube and suspended in MeNH2 solution (1 ml). The mixture was heated to 65 °C for 10 min, cooled to room temperature (water/icebath) and centrifuged for 1 min (10,000 g). The supernatant was separated from the CPG beads, the beads were washed with RNase free water (2 × 0.25 ml), all supernatants were combined and dried (2 h under nitrogen stream, then freeze dried). The oligoribonucleotide was resuspended in anhydrous NEt3·3HF/NEt3/NMP solution (250 µl of a solution of 1.5 ml NMP, 750 μlNEt3 and 1.0 ml NEt3·3HF), heated to 65 °C for 1.5 h, cooled to room temperature and quenched with 3M NaOAc solution (25 µl). n-BuOH (1 ml) was added to the mixture, which was then thoroughly mixed, cooled to −70 °C for 1 – 2 h to encourage further precipitation and centrifuged for 30 min (4 °C, 13 000 g). The supernatant was removed, the pellet washed with 70% EtOH (2 × 500 µl) and then dried in vacuo (30 min). The dry precipitate was dissolved in RNase free water (1 ml) and desalted using a Nap™-10 column following the standard protocol. The resulting solution was freeze dried over night leaving the oligoribonucleotide as a white foam/powder. Samples for MALDI-mass spectrometry were prepared as follows36: Dowex™ ion-exchange beads were rigorously cleaned with dilute HCl, washed with water, then treated with dilute NH3 and finally washed with water again to generate Dowex-NH4+. Diammonium citrate (DAC) (100 mg) was dissolved in water (1 ml) and HPA (34.8 mg) was dissolved in 1:1 acetonitrile/water (1 ml). The HPA solution was filtered through Dowex-NH4+ and the DAC solution (100 µl) was added to prepare the matrix stock. Prior to MALDI-MS acquisition, matrix stock (20 µl) was mixed with Dowex-NH4+ (5 µl) and each oligoribonucleotide sample (1 µl) was mixed with Dowex-NH4+ (19 µl). After 30 min the matrix and after drying, the sample solution (0.5µl) were spotted onto the sample well and allowed to dry prior to confirmatory analysis by MALDI-MS.
RNA purification
Total RNA was prepared from mouse (C57BL/6) tissues using Trizol reagent (Invitrogen). The poly(A) RNA was purified twice using oligo(dT) cellulose (Fluka) followed by oligo(dT) magnetic beads (Invitrogen). All samples were quantified and assayed for poly(A) purity using an Agilent Bioanalyser.
Analysis of gene-specific mRNA cap structures
Regions corresponding to the first 300 to 500 nucleotides of the target messages were PCR amplified from cDNAs using the following oligonucleotides:
Apoa1forward GCTCCGGGGAGGTCACCCACACCT and
Apoa1reverse CAATGGGCCCAGCCGTTCCTGCAGC;
Albforward CCCCACTAGCCTCTGGCAAAATGAAGTG and
Albreverse GGCTGGGGTTGTCATCTTTGTGTTGCAG;
Prm2forward GCTGGGTGTGCGCGAGTCAGGGGCTC and
Prm2reverse CTTGTGGATCCTATGTAGCCTCTTACG;
Pabpc1forward CGGCGGTTAGTGCTGAGAGTGCGGAG and Pabpc1reverse GAAGTTCACGTACGCGTAGCCCAAGG. Prior to amplification, the forward oligonucleotides were 5’ phosphorylated, the amplification products were subsequently digested with lambda nuclease (New England BioLabs) to leave the single stranded antisense DNA strand. 100 ng of this ssDNA (2 µl) was spotted onto 2 mm × 2 mm teeth cut from a Hybond N+ membrane ( Amersham) (Fig. 2B,C) and UV cross-linked (Stratalinker). Membranes were prehybridised at 42 °C in 40 % formamide with 5 × Denhardt’s, 3 % SDS, 0.3 M NaCl, 50 mM sodium phosphate buffer (pH 7.0) and 0.1 mg ml−1 sonicated salmon sperm DNA.
600 ng of poly(A) RNA was digested with 20 units of Tobacco Acid Pyrophosphatase (Epicentre) for 30 minutes at 37 °C. The 5’ phosphate of the exposed cap adjacent nucleotide was removed by the addition of 10 units of Alkaline Phosphatase (Fermentas) and incubation for a further 15 minutes at 37 °C. After phenol-chloroform extraction and ethanol precipitation, RNA samples were resuspended in 20 µl of sterile distilled water and 5’ ends were labelled using 30 units T4 polynucleotide kinase (PNK, Fermentas) and 7.4 MBq [γ-32P] ATP at 37 °C for 30 minutes. The PNK was heat inactivated (70 °C for 15 min) and the reaction made up to 60 µl with sterile distilled water then passed through a P-30 spin column (Bio-Rad) to remove unincorporated isotope. A 1 µl aliquot was taken, added to 9 μl of nuclease P1 buffer and digested with P1 (Sigma) for one hour at 37 °C. 1.5 μl of the released 5’ monophosphates from this digest was then analysed by 2D TLC as described previously25. The remaining end labelled RNA was fragmented to lengths of approximately 120 nt by the addition of Na2CO3 to a final concentration of 60 mM and NaHCO3 to a final concentration of 40 mM followed by incubation for one hour at 60 °C.12 µl 3M sodium acetate pH 5.2 was then added and the RNA precipitated with ethanol. The pellet was resuspended in 200 µl of pre-hybridisation buffer then added to the membranes (final volume 2 ml) and hybridised overnight at 42 °C. Membrane washings were carried out with 2 ×, 1 × and 0.2 × SSC, 0.1% SDS. Two final washes were carried out at 60 °C, with 0.2 × SSC but with the SDS omitted. Hybridised membranes were exposed to storage phosphor screens (K-screen; KODAK) and imaged using Bio-Rad Molecular Imager FX in combination with Quantity One 4.6.3 software (Bio-Rad). Individual teeth containing the target mRNAs end labelled at the cap adjacent position were cut off and digested with P1 nuclease (Sigma) in a final volume of 3 µl. All of this digestion mix was applied to a cellulose coated TLC plate (20 × 20 cm, MERCK) and developed as described previously25.
References
Langberg, S. R. and Moss, B. Post-transcriptional modifications of mRNA. Purification and characterization of cap I and cap II RNA (nucleoside-2’-)-methyltransferases from HeLa cells. J. Biol. Chem., 256, 54–60 (1981).
Bélanger, F., Stepinski, J., Darzynkiewicz, E. and Pelletier, J. Characterization of hMTr1, a Human Cap1 2‘-O-Ribose Methyltransferase . J. Biol. Chem. 285, 33037–33044 (2010).
Werner, M., et al. 2’-O-ribose methylation of cap2 in human: function and evolution in a horizontal mobile family. Nucl. Acids Res., 39, 4756–4768 (2011).
Wei, C. M., Gershowitz, A. and Moss, B. N6,O2 -dimethyladenosine a novel ribonucleoside next to the 5’terminal of animal cell and virus mRNAs. Nature, 257, 251–253 (1975).
Keith, J. M., Ensinger, M. J. and Moss, B. HeLa cell RNA(2’-O-methyladenosine –N6-)-methyltransferase specific for the capped 5’-end of messenger RNA. J. Biol. Chem., 253, 5033–5041 (1978).
Juven-Gershon, T., Hsu, J-Y., Theisen, J. W. M. and Kadonaga, J. T. The RNA polymerase II core promoter – the gateway to transcription. Curr. Opin. Cell Biol., 20, 253–259 (2008).
Itaya, T., Tanaka, F. and Fujii, T. Purines 6. Rate study and mechanism of Dimroth rearrangement of 1-alkoxy-9-alkyladenines and 1-alkyl-9-methyladenines. Tetrahedron, 28, 535–547 (1972).
Fujii, T., et al. Purines 29. Synthesis of 9-alkyl-2-deuterio-N6-methoxyadenines and 2-deuterio-N6,9-dimethyladenine – tautomerism in 9-substituted N6-alkoxyadenines. Chem. Pharm. Bull., 35, 4482–4493 (1987).
Kruger, T., Wagner, C., Bruhn, C., Lis, T. and Steinborn, D. Molecular and crystal structures of dialkylated adenines (N-6,N-9-Me(2)Ade, N-3,N-6-MeBnAde) and cytosines (N-1,N-4-Me(2)Cyt). J. Mol. Struc., 891, 110–114 (2008).
Höbartner, C., et al. The synthesis of 2 '-O-[(triisopropylsilyl)oxy] methyl (TOM) phosphoramidites of methylated ribonucleosides m(1)G, m(2)G, m(2)(2)G, m(1)I, m(3)U, m(4)C, m(6)A, m(2)(6)A) for use in automated RNA solid-phase synthesis. Monatsh. Chem., 134, 851–873 (2003).
Veliz, E. A., Easterwood, L. M. and Beal, P. A Substrate analogues for an RNA-editing adenosine deaminase: Mechanistic investigation and inhibitor design. J. Am. Chem. Soc., 125, 10867–10876 (2003).
Porcher, S. and Pitsch, S. Synthesis of 2 '-O-[(triisopropylsilyl)oxy]methyl ( = tom)-protected ribonucleoside phosphoramidites containing various nucleobase analogues. Helv. Chim. Acta, 88, 2683–2704 (2005).
Dai, Q., et al. Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N-6-methyladenosine. Nucl. Acids Res., 35, 6322–6329 (2007).
Izaurralde, E., et al. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell, 78, 657–668 (1994).
Flaherty, S. M., Fortes, P., Izaurralde, E., Mattaj, I. W. and Gilmartin, G. M. Participation of the nuclear cap binding complex in pre-mRNA 30 processing. Proc. Natl. Acad. Sci. U. S. A., 94, 1893–11898 (1997).
Hernádez, G., Altmann, M. and Lasko, P. Origins and evolution of the mechanisms regulating translation initiation in eukaryotes. Trends Biochem. Scis, 35, 63–73 (2009).
Muthukrishnan, S., Morgan, M., Banerjee, A. K. and Shatkin, A. J. Influence of 5’-terminal m7G and 2’-O-methylated residues on messenger ribonucleic acid binding to ribosomes. Biochemistry, 15, 5761–5768 (1976).
Kuge, H., Brownlee, G. G., Gershon, P. D. and Richter, J. D. Cap ribose methylation of c-mos mRNA stimulates translation and oocyte maturation in Xenopus laevis . Nucl. Acids Res., 26, 3208–3214 (1998).
Meis, R. and Meis, J. E. Improve the translation efficiency of any 5’-capped mRNA with the new ScriptCapTM 2’-O-methyltransferase. EPICENTRE Forum, 13, 5–6 (2006).
Bouvet, M., et al. In vitro reconstruction of SARS-coronavirus cap methylation. PLoS Pathog., 6, e1000863 (2010).
Lockless, S. W., Cheng, H. T., Hodel, A. E., Quiocho, F. A. and Gershon, P. D. Recognition of capped RNA substrates by VP39, the vaccinia virus-encoded mRNA cap-specific 2 '-O-methyltransferase. Biochemistry, 37, 8564–8574 (1998).
Daffis, S., et al. 2’-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature, 468, 452–456 (2010).
Züst, R., et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nature Immunol., 12, 137–144 (2011).
Banerjee, A. K. 5-Terminal cap structures in eukaryotic messenger ribonucleic acids. Microbiol. Reviews, 44, 175–205 (1980).
Zhong, S., et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. The Plant Cell, 20, 1278–1288 (2008).
Wei, C. M., Gershowitz, A. and Moss, B. 5’-terminal and internal methylated nucleotide-sequences in Hela-cell messenger-RNA. Biochemistry, 15, 397–401 (1976).
Clancy, M. J., Shambaugh, M. E., Timpte, C. S. and Bokar, J. A. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6 -methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucl. Acids Res.., 30, 4509–4518 (2002).
Kane, S. E. and Beemon, K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol. Cell. Biol., 5, 2298–2306 (1985).
Nichols, J. L. N6-methyladenosine in maize poly(A)-containing RNA. Plant Sci. Letters, 15, 357–361 (1979).
Aritomo, K., Wada, T. and Sekine, M. Alkylation of 6- N -acylated adenosine derivatives by the use of phase transfer catalysis. J. Chem. Soc. Perkin Trans., I, 1837 (1995).
Kimura, M., Ishida, K., Kashiwabara, S. I. and Baba, T. Characterization of two cytoplasmic poly(A)-binding proteins, PABPC1 and PABPC2, in mouse spermatogenic cells. Biol. Reprod., 80, 545–554 (2009).
Frith, M. C., Valen, E., Krogh, A., Hayahizaki, Y., Carninci, P. and Sandelin, A. A code for transcription initiation in mammalian genomes. Genome Res., 18, 1–12 (2008).
Bokar, J. A., Shambaugh, M. E., Polayes, D., Matera, A. G. and Rottman, F. M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6 -adenosine)-methyltransferase. RNA, 3, 1233–1247 (1997).
Niedzwiecka, A., et al. Biophysical studies of eIF4Ecap-binding protein: recognition of mRNA 5’ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J. Mol. Biol., 319, 615–635 (2002).
Tomoo, K., et al. Crystal structures of 7-methylguanosine 5‘-triphosphate (m(7)GTP)- and P-1-7-methylguanosine-P-3-adenosine-5‘,5‘-triphosphate (m7GpppA)-bound human full-length eukaryotic initiation factor 4E: biological importance of the C-terminal flexible region Biochem . Journal, 362, 539–544 (2002).
Langley, G. J., Herniman, J. M., Davies, N. L. and Brown, T. Simplified sample preparation for the analysis of oligonucleotides by matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom., 13, 1717–1723 (1999).
Acknowledgements
Z. Bodi was supported by a BBSRC Daphne Jackson Fellowship and a Welcome Trust VIP award, J. Button was supported by a BBSRC Studentship.
Author information
Authors and Affiliations
Contributions
SK and CH developed and carried out the novel chemical synthesis of phosphoramidites. SZ, JB and RF developed the cap labeling protocols. ZB and RF wrote the manuscript. MA provided materials and useful comments. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kruse, S., Zhong, S., Bodi, Z. et al. A novel synthesis and detection method for cap-associated adenosine modifications in mouse mRNA. Sci Rep 1, 126 (2011). https://doi.org/10.1038/srep00126
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00126
This article is cited by
-
Dynamic regulation of N6,2′-O-dimethyladenosine (m6Am) in obesity
Nature Communications (2021)
-
FTO controls reversible m6Am RNA methylation during snRNA biogenesis
Nature Chemical Biology (2019)
-
Distinguishing RNA modifications from noise in epitranscriptome maps
Nature Chemical Biology (2018)
-
Reversible methylation of m6Am in the 5′ cap controls mRNA stability
Nature (2017)
-
The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells
Nature Medicine (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
