
Abstract
Tropical coral reefs are among the most affected ecosystems by climate change and face increasing loss in the coming decades. Effective conservation strategies that maximize ecosystem resilience must be informed by the accurate characterization of extant genetic diversity and population structure together with an understanding of the adaptive potential of keystone species. Here we analyzed samples from the Tara Pacific Expedition (2016–2018) that completed an 18,000 km longitudinal transect of the Pacific Ocean sampling three widespread corals—Pocillopora meandrina, Porites lobata, and Millepora cf. platyphylla—across 33 sites from 11 islands. Using deep metagenomic sequencing of 269 colonies in conjunction with morphological analyses and climate variability data, we can show that despite a targeted sampling the transect encompasses multiple cryptic species. These species exhibit disparate biogeographic patterns and, most importantly, distinct evolutionary patterns in identical environmental regimes. Our findings demonstrate on a basin scale that evolutionary trajectories are species-specific and can only in part be predicted from the environment. This highlights that conservation strategies must integrate multi-species investigations to discern the distinct genomic footprints shaped by selection as well as the genetic potential for adaptive change.
Similar content being viewed by others

Introduction
Coral reef ecosystems harbor approximately one-third of the world’s multicellular marine biodiversity1 despite covering only ~0.1% of the seafloor2. Critically, however, these ecosystems are also some of the most sensitive to climate change3 with decreasing global coral coverage over recent decades4. In line with even moderate projections of global warming, 70–90% of coral reefs may disappear in the coming decades5,6, jeopardizing the biological diversity they support and the more than 500 million people who rely on the services they provide7. To minimize further losses, and maximize their potential for recovery, the implementation of effective conservation strategies for these ecosystems is imperative8,9,10. Moreover, developing adaptation strategies and building increased reef resilience based on the natural adaptive potential of corals has become a requirement to ensure a future for coral reefs5,9.
A critical component underlying the success of these conservation strategies is the preservation of the reef-building corals that form the ecological and physical foundations of reef ecosystems8,11. Accurately characterizing the extant genetic diversity and genetic structure of these corals12, as determined by their connectivity12,13,14, is an essential prerequisite for planning marine reserve networks that can replenish lost genetic diversity8. Characterization of life history traits such as growth form and reproductive mode among other biological attributes such as acclimation and adaptation potential are also essential, as different species may have significantly disparate responses to prevailing and historical environmental conditions15,16.
However, unrecognized interspecific (cryptic) diversity and high intraspecific morphological plasticity, e.g., in the genera Acropora and Pocillopora17,18,19 make characterizing genetic diversity through morphological characteristics alone problematic. The accurate characterization of diversity is improved through integration of morphological data with genome-wide sequencing strategies, e.g., RAD-Seq, RNA-Seq, whole-genome/-exome shotgun sequencing16,20 or, more recently, target-capture of ultraconserved elements (UCE)21,22,23,24. Such approaches enable fine-scale resolution of genetic lineages, estimations of relatedness and divergence timings, and identification of genomic loci under selection25, all of which are of high value to conservation planning efforts. To best inform conservation efforts, robust genetic characterization of corals (i.e., from deep whole-genome sequencing) across large geographic ranges are therefore required. Such campaigns are however difficult to achieve due to the logistical challenge of standardized sample collection and considerable financial requirements.
Across the Pacific Ocean it is largely unknown how coral diversity is structured, how the prevailing environment has shaped evolutionary history, and whether the consequential evolutionary trajectories are shared between corals. To begin to answer these questions, we analyzed samples from the Tara Pacific Expedition26 that ran from 2016 to 2018 and completed an 18,000 km longitudinal transect of the Pacific Ocean, sampling three widespread corals, two scleractinian corals—Pocillopora meandrina and Porites lobata—and one hydrozoan coral—Millepora cf. platyphylla—across 33 sites from 11 islands. These species were targeted using their colony morphologies which inevitably led to the sampling of cryptic species widespread in corals. We conducted ultra-deep metagenomic sequencing of 269 coral colonies. This sequencing effort was then combined with morphological analyses and climate data to identify the presence of putative cryptic species, determine the standing genetic diversity and population structure within each characterized lineage, and reveal disparate patterns of environmentally linked genomic loci under selection. Our work demonstrates that different species are differentially shaped by the same prevailing environment and exhibit distinct genomic footprints. Such information is critical to guide ongoing and future conservation efforts.
Results
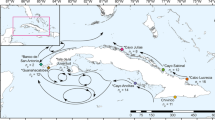
Morphology-guided sampling of colonies resembling P. meandrina, P. lobata, and Millepora cf. platyphylla resulted in the collection of 106, 109, and 54 coral colonies across 11 islands, spanning 18,000 km of overwater distance (Fig. 1 and Supplementary Table 1). To resolve species identities, we designated the sampling regime as morphological-based primary species hypotheses (PSH) that we further developed into secondary species hypotheses (SSH) through an integrated taxonomic approach by analyzing genetic diversity and morphological parameters of the sampled colonies14. For each of the three coral genera, building on the genomic and transcriptomic sequences produced by the Tara Pacific consortium27, we used multiple genetic analyses to characterize the extent and distribution of extant genetic diversity.

The number of samples belonging to a given genetic delineation are given for each of the genera. Reference reefs from the United Nations Environment Programme—World Conservation Monitoring Center’s (UNEP-WCMP) Global Distribution of Coral Reefs dataset111,112 are plotted in red using reefMapMaker113.
Genetic delineation and biogeography of Pocillopora spp.
For Pocillopora, Maximum Likelihood (ML; Supplementary Fig. 1) and SVDquartet (used to estimate the species-tree topology based on SNP data under a multi-species coalescent model; Fig. 2a) trees shared a similar topology with five well-supported clades (SVD1-SVD5) containing the same constituent samples and the same single outlier sample (I09S03C010). The SVD1 clade was more distantly related to the other SVD clades and we could observe two pairs of sister clades: SVD3 & SVD5 and SVD2 & SVD4. These clades were further supported through the estimation of individual ancestry coefficients using sNMF genetic clustering and principal component analysis (PCA), which predicted five ancestral populations (K1-K5). While minimal introgression was apparent across most samples, several subsets of samples from specific islands, resolved to specific subclades of the SVD tree, displayed higher levels of introgression (e.g., K3_I15, K4_I05, and K5_I07; Fig. 2c). The one sample that grouped as an outlier in the ML and SVDquartet trees demonstrated a hybrid ancestry with a ~50/50 split of the ancestral populations otherwise associated with samples from the SVD2 and SVD3 groupings. Despite the island-specific patterns of admixture in some lineages, sNMF analysis within the identified SVD lineages identified no further genetic clustering. The SVD groupings were recapitulated in the PCA across the four highest-scoring principal components (PCs; Fig. 2b). We tested multiple SSHs by coalescence analysis using two replicate runs of BFD* with samples categorized by SVD group. The most likely species hypothesis differed between runs, but the second most likely hypothesis in both runs designated each SVD a separate species (Supplementary Table 2). We adopted this 5-species SSH as our working hypothesis. Of note, the difficulty in such assignments lies in avoiding mixing of isolated populations and species. Given that one has to drastically reduce the sampling size for this type of analysis, we chose to discard SSHs whose likelihood ranks varied between replicate runs. To assign species names to our SSH, two representatives for each SVD grouping were mapped onto the Pocillopora reference target-capture sequences from a previous study28 to then call the species-diagnostic SNPs generated therein. While SVD1-SVD4 could be assigned to the following species names: Pocillopora cf. effusa (SVD1), Pocillopora meandrina (SVD2), Pocillopora verrucosa (SVD3), and Pocillopora grandis (SVD4), SVD5 was most similar to a lineage with no formal name that is closely related to P. verrucosa (hereafter referred to as SSH5_pver; Supplementary Fig. 2). SVD1 was attributed the name P. cf. effusa, as P. effusa still lacks a formal taxonomic description. In addition, the species name attribution of each genetic lineage was further confirmed using mtORF sequences extracted from our metagenomic data and compared to previously published records19,29,30,31 (see Supplementary Table 3 and Supplementary Fig. 3). When assessing how samples from the different species were distributed across our sampling regime, we found that species exhibited region-specific distributions with each region composed of adjacent islands (Fig. 1). P. meandrina and P. verrucosa were distributed in the western Pacific, P. cf. effusa and SSH5_pver in the central Pacific, and P. grandis in the eastern and central Pacific. At all but the four most eastern sites (Easter, Malpelo, Coiba, and Las Perlas), multiple species were found in sympatry (Fig. 1). Notably however, P. verrucosa and SSH5_pver as well as P. meandrina and P. grandis, the two most genetically similar species pairings, were only found in allopatry in our samples.

a SVDquartet tree. Node support ≥80% from 100 bootstraps is indicated with a black dot. Samples are grouped into five clades (SVD1-SVD5) based on tree morphology and sNMF analysis which form the secondary species hypotheses (SSHs). Species names resulting from subsequent analyses (see the relevant section of this study) have been annotated. Samples not included in the sNMF analysis (due to removal of clonal/multilocus lineages) are underlined. b Principal component analysis (PCA) based on genome-wide SNPs. The relative eigenvalues are shown in the subplots with the displayed eigenvectors shaded black. c Hierarchical genetic clustering using sNMF. Bars in each column represent the contribution of predicted ancestral lineages to each sample. Samples are ordered longitudinally by island.
Genetic delineation and biogeography of Porites spp.
The ML (Supplementary Fig. 1) and SVDquartet (Fig. 3a) trees generated for Porites both supported the existence of three clades (SVD1-SVD3) with SVD2 and SVD3 more closely related to each other than SVD1. All samples resolved within one of these clades with the exception of sample I15S02C011 that resolved between SVD1 and the other two clades. These clades were further supported by sNMF genetic clustering and principal component analysis (PCA). The sNMF analysis suggested three ancestral populations (K1-K3) with minimal introgression apparent (Fig. 3c). Notably, there was exact agreement between SVDquartet clade placement and sNMF assignments (according to highest admixture coefficient; hereafter, SVD1 will be related to K1, SVD2 to K2, SVD3 to K3) for all but three of the samples, which exhibited significant levels of admixture (see Supplementary Table 4). Unlike in Pocillopora, genetic subclustering was detected within each of the sNMF groupings with two, three, and four ancestral populations predicted for K1, K2, and K3 (hereafter referred to as K1a, K1b, K2a, etc.; Fig. 3c), respectively. These subclusterings matched the subclade structure in the SVD groupings (exact match in SVD1 and SVD3, close match in SVD2; Fig. 3a) and largely demonstrated island-specific, but not endemic, patterns (Fig. 1). We tested the SSHs by coalescence analysis using the sNMF subcluster groupings (i.e., K1a, K1b, K2a, etc.). As in Pocillopora, the most likely species hypothesis differed between runs, but the second most likely hypothesis in both runs designated K1, K2 and K3 as distinct species. We adopted this 3-species SSH as our working hypothesis and hereafter refer to SSH1-SSH3 in reference to K1-K3. By mapping our metagenomic reads on species-diagnostic loci32, we were able to assign SSH1 to Porites evermanni and SSH2 and SSH3 to P. lobata (Supplementary Fig. 4). Thus, SSH2 and SSH3 putatively constitute two cryptic lineages within the P. lobata species. We will therefore refer to them hereafter as SSH2_plob and SSH3_plob. As for Pocillopora, each of the SSHs were distributed across multiple islands with one or two lineages found at each island. SSH2_plob and SSH3_plob were most widely distributed with each lineage found at seven islands (absent in only Malpelo and Las Perlas), while P. evermanni (SSH1) was found only at the easternmost three sites (Malpelo, Coiba, and Las Perlas; side-by-side with SSH2_plob; Fig. 1). In contrast to Pocillopora where genetically similar species pairings were only found in allopatry, the more genetically similar species SSH2_plob and SSH3_plob were found in widespread sympatry.

a SVDquartet tree. Node support ≥80% from 100 bootstraps is indicated with a black dot. Samples are grouped into three main clades (K1-K3) and two, three, and four subclades, respectively (K1a, K1b, K2a, etc.) based on tree morphology and sNMF analysis with the main clades representing the three secondary species hypotheses (SSHs; annotated). b Principal component analysis (PCA) based on genome-wide SNPs. The relative eigenvalues are shown in the subplots with the displayed eigenvectors shaded in black. c Hierarchical genetic clustering using sNMF across all samples with main clades below and subclades above. Bars in each column represent the contribution of predicted ancestral lineages to each sample. Samples are ordered by cluster and then longitudinally by island.
Genetic delineation and biogeography of Millepora spp.
In Millepora, agreement in topology between the ML (Supplementary Fig. 1) and SVDquartet (Fig. 4a) trees was poor, unlike in Pocillopora and Porites. While the SVD tree showed relatively strong bootstrap values that supported the existence of six clades (SVD1-SVD6), the branches of the ML tree lacked support and distinct clades were difficult to resolve due to the low number of SNPs identified, with the exception of the M. intricata samples (identified as a distinct species based on field-based morphological assessment; see Methods) that were resolved separately from all other samples in both trees. The sNMF analysis predicted four ancestral populations (K1-K4) with levels of introgression varying considerably. Unlike in Pocillopora and Porites, the sNMF ancestral populations did not correspond directly to the SVD groupings, and rather, discrete combinations of the four ancestral populations resolved six groups that corresponded to the SVD clades (Fig. 4c). PC1 of the PCA analysis grouped the M. intricata samples separately from all others, while the remaining samples grouped according to the five remaining SVD groups across PCs 2–4 (Fig. 4b). Due to the low levels of support in the ML tree, the higher levels of introgression from predicted ancestral populations in extant samples (sNMF analysis), and the lack of direct correspondence between the sNMF ancestral populations and the SVD groupings, we considered the five non-M. intricata SVD groupings (SVD2-SVD6) to be representative of a single species, designated as Millepora cf. platyphylla33. Unlike in Pocillopora and Porites, Millepora SVD clades were all endemic to specific islands with the exception of SVD6. Nevertheless, a split at the basal node of the SVD6 group partitioned the samples into two clades with all members endemic to either Gambier or Aitutaki (Figs. 1 and 4a).

a SVDquartet tree. Node support ≥80% from 100 bootstraps is indicated with a black dot. Samples are grouped into six clades (SVD1-SVD6) based on tree morphology and sNMF analysis. Samples not included in the sNMF analysis (due to the removal of clonal/multilocus lineages) are underlined. b Principal component analysis (PCA) based on transcriptome-wide SNPs. The relative eigenvalues are shown in the subplots with the displayed eigenvectors shaded black. c Hierarchical genetic clustering using sNMF. Bars in each row represent the contribution of predicted ancestral lineages to each sample. Samples are ordered longitudinally by island.
Morphological analysis
In parallel to the genetic analyses, we also conducted morphological analyses that employed a multivariate suite of macromorphological measurements (see “Methods”). This served a dual purpose: first, we wanted to assess whether we could identify morphological variation between populations, and second, we wanted to assert whether such variation corresponded to the genetically identified SSHs, to provide genetic and morphological evidence of the species designations. Morphological analysis revealed structured variation in Pocillopora (Supplementary Fig. 5), Porites (Supplementary Fig. 6), and Millepora (Supplementary Fig. 7). We also found significant PERMANOVA results when testing the multivariate morphological characteristics against the genetic delineations, which demonstrated that some of the variation could be explained by the SSH designations (Pocillopora: F1,75 = 1.6669, P = 0.013, n = 77, Porites: F2,78 = 2.3329, n = 81, P = 0.001, and Millepora: F4,45 = 1.5601, P = 0.003, n = 50). Critically however, for each of the coral genera, there was no combination of morphological features that could identify the genetic resolutions; rather, each of the morphotype categorizations contained multiple SSHs. Within-genus pairwise PERMANOVA testing identified differences between all pairings except for P. meandrina vs. P. verrucosa and P. meandrina vs. SSH5_pver in Pocillopora, and SSH6 vs. SSH5, SSH6 vs. SSH2, SSH4 vs. SSH3, SSH4 vs. SSH2, SSH5 vs. SSH3, and SSH3 vs. SSH2 in Millepora. All within-Porites pairings returned significant results.
Evolutionary history of among- and within-species genetic differentiation
To further resolve evolutionary history in the Pocillopora and Porites species, we investigated introgression and pairwise divergence for the SSHs (Fig. 5). For Millepora, an insufficient number of SNPs and representative samples meant that these analyses were not conducted. In Pocillopora, three admixture events were predicted and verified based on significant results in the relevant f4 indices34 (Fig. 5b, c) where two of the admixture events were from ancestral nodes to child clades and the third was inter-SSH from P. verrucosa_I06 to P. meandrina_I09. The resulting TreeMix consensus tree showed a similar topology to that of the SVD tree with those groups from P. effusa more differentiated from the other SVD groups and with two pairs of sister clades formed by P. meandrina & P. grandis and P. verrucosa & SSH5_pver (Fig. 5b). After accounting for the three introgression events, residuals (obtained from fitting the tree model to the observed data) indicated potential remaining admixture within each of the five species (Fig. 5a). Similar to the Treemix tree, the f2 values (Supplementary Fig. 8A) and the distributions of genomic Weir’s FST values computed from 500 bp sliding windows (Supplementary Fig. 8B) recapitulated the high divergence of P. effusa from the other more genetically similar two species pairs (P. meandrina & P. grandis and P. verrucosa & SSH5_pver). Taken together, these findings reinforce the pattern in Pocillopora that the genetically closely related sister clade species pairings (P. meandrina & P. grandis and P. verrucosa & SSH5_pver) are found in our samples in allopatry rather than sympatry. In Porites, three admixture events were predicted and confirmed (f4 significant results; Fig. 5d, e) with one within-SSH, one inter-SSH, and one from an ancestral to a child clade. As for Pocillopora, the resulting TreeMix consensus tree topology agreed with that of the SVD tree, with P. evermanni (SSH1) being more differentiated compared to SSH2_plob and SSH3_plob. After accounting for the three admixture events, residuals indicated potential remaining admixture within each of the three SSHs and also between SSH2_plob and SSH3_plob (Fig. 5d). The f2 values (Supplementary Fig. 8C) and the distributions of Weir’s FST from 500 bp sliding windows (Supplementary Fig. 8D) both supported the relative divergence of P. evermanni (SSH1) from the other two SSHs, and in contrast to Pocillopora, it also showed a correlation between sympatry and genetic relatedness.

a, d residuals indicating potential remaining admixture between samples in Pocillopora and Porites, respectively, after accounting for the three migration events annotated in the TreeMix tree. b, e Treemix consensus trees with migration events and their weights annotated. c, f Corresponding f4 indices.
Environment as a predictor of genetic lineage distribution
The gradientForest algorithm35 was used to assess whether parameters related to past sea surface temperature (SST) and depth36 are predictors for the geographic distribution of the genetic lineages determined for Pocillopora and Porites (Supplementary Fig. 8). For both genera, the geographic distribution of all lineages could be partially predicted based on past SST and depth with varying degrees of accuracy (Pocillopora: P. cf effusa/SVD1: R2 = 0.161, P. meandrina/SVD2: R2 = 0.412, P. verrucosa/SVD3: R2 = 0.348, P. grandis/SVD4: R2 = 0.565; Porites: P. evermanni/SSH1: R2 = 0.687, SSH2_plob: R2 = 0.290, SSH3_plob: R2 = 0.513). Among the three most contributing predictors for the geographic distribution of Pocillopora lineages, two were related to cold spells, namely the number of days and the mean number of days per year between 2002 and the sampling date where the SST dropped 1 °C or more below the weekly (long-term) climatology (TSA_cold_freq_sum_day: R2 = 0.095 and TSA_cold_freq_mean_day: R2 = 0.0889). The third predictor was related to SST anomalies, i.e. the maximum number of days within 1 year between 2002 and the sampling date where the SST was 1 °C or more higher than weekly climatologies (SST_anomaly_freq_max_day: R2 = 0.090). The geographic distribution of Porites was better predicted by SST anomalies, namely the mean number of days per year between 2002 and the sampling date where SST was 1 °C or more higher than weekly climatologies (SST_anomaly_freq_mean_day: R2 = 0.112), the mean SST anomaly (in °C) per year between 2002 and sampling date (SST_anomaly_mean_DegC: R2 = 0.095), and the seasonal mean SST (seasonal_mean_DegC: R2 = 0.095). Depth of the sampling sites was only a minor predictor for the geographic distribution of species within both genera (Pocillopora: R2 = 0.034, predictor 57/68, and Porites: R2 = 0.023, predictor 46/68).
Correlation of genetic relatedness with geographic distance and climate
To assess for isolation-by-distance and a possible effect of temperature regime on the evolutionary trajectories of species, we separately tested for correlation between genetic distance (encoded as pairwise FST/(1-FST) distances generated from pairwise group FST distances; see “Methods”) and both, geographical distance (calculated as the overwater distance between islands) and climate (represented by past temperature difference based on island-wise Euclidean past temperature distances generated from a reduced dimensionality representation; see “Methods”)36. In Pocillopora, separate correlation analyses of genetic distance against geographical distance and temperature difference returned nonsignificant results across and within species (Supplementary Table 5). By contrast, significant results were returned for Porites across all species (geography, r = 0.397, P = 0.005; temperature, r = 0.361, P = 0.020) and within SSH2 (geography, r = 0.585, P = 0.036; temperature, r = 0.641, P = 0.010) and SSH3 (geography only, r = 0.591, P = 0.008). In Millepora, a significant result was observed for all lineages against temperature (r = 0.930, P = 0.019) but not geography (r = 0.722, P = 0.053). For each coral genus, geographical distance significantly correlated with past temperature distance (Supplementary Table 5).
Influence of temperature regime on evolutionary trajectories
To investigate whether temperature (as a prevalent stressor of the coral metaorganism) was a potential driver of species divergence patterns in Pocillopora and Porites, we assessed whether SNPs that aligned with past temperature regimes exist. Indeed, we could identify SNPs in Pocillopora and Porites whose local allelic frequencies could be predicted by past temperature regime (i.e., temperature outlier SNPs; Pocillopora— 38,229 out of 461,989 SNPs (8.27%); Porites—6441 out of 57,966 SNPs (11.11%); at RDA q value <0.1 from the unlinked dataset, see “Methods”; Fig. 6). To determine those SNPs potentially involved in species divergence patterns, we queried which of the SNPs originated from a set of genomic islands of differentiation (GID) defined as the 1% of the 500 bp bins with the highest mean FST value among SSHs for Pocillopora and Porites. In Pocillopora, 20 of the top 100 temperature outlier SNPs (ranked by P value) were located in these GIDs (Fig. 6). By contrast, Porites had only 3 such SNPs in GIDs, suggesting that species differentiation is more closely correlated to thermal history in Pocillopora than in Porites. We also investigated whether this difference could be due to drift or actively maintained due to divergent adaptation. In Pocillopora, 21% of the whole genome was predicted to be under divergent selection (at the level of between-SSH comparisons), a considerably higher proportion than the 7% predicted in Porites. However, independent of this baseline difference between the two genera, in Pocillopora 41% of SNPs in the top 100 temperature outliers were predicted to be under selection, and this proportion grew to 61% (12 out of 20) when considering those outlier SNPs under selection and residing in GIDs, approximately double and triple that of the proportion of SNPs under divergent selection across the whole genome (21%). By contrast, in Porites a decrease from the 7% of SNPs under divergent selection across the whole genome was observed when considering either those outlier SNPs predicted to be under selection (2%) or those under selection and residing in GIDs (0%; 0 out of 3). This suggests that while the differentiation and distribution of the Pocillopora species are due to and/or maintained by divergent selection driven by temperature regimes, in Porites divergence among SSHs is likely due to either drift or other environmental factors (of note, many environmental factors correlate with temperature).

a Pocillopora, b Porites. The x axis gives cumulative position in the genome with contigs sorted by decreasing size order. P values used to generate the y axis are from the RDA analysis. Circles: SNPs not included in genomic islands of differentiation between SSHs (GID); triangles: SNPs included in GIDs; dark gray: SNPs not significantly associated to past temperature variation; light gray: SNPs significantly associated to temperature variation; gold: top 100 temperature outlier SNPs; red: SNPs among the top 100 temperature outliers under divergent selection among species according to Flink analysis.
Discussion
Our primary species hypothesis of one species per coral genus, based on in situ morphological assessment (i.e., during underwater collection), was genetically validated only for Millepora cf. platyphylla. The corals collected and putatively designated as Pocillopora meandrina and Porites lobata produced SSHs of 5 and 3 species, respectively, that were supported by multiple genetic analyses (Figs. 2–5 and Supplementary Fig. 8). Subsequent morphological analyses based on underwater colony pictures of the corals resolved a number of morphotypes (Supplementary Figs. 5–7) that showed correlation with the genetically resolved lineages in all three genera. However, multiple genetic lineages were associated with all designated morphotypes and the identification of lineage-diagnostic morphological features was not possible based on underwater colony pictures. Our finding underlines the difficulty of defining morphological criteria that can be readily derived from photographs and therefore supports the well-established existence of high morphological plasticity in corals, as exemplified by the genus Pocillopora37,38,39. Further, it reinforces that plasticity is a confounding factor to achieve effective taxonomic resolutions37,40,41 and identification of species in the field. Finally, it highlights the difficulties in conducting large geographic surveys on targeted coral species due to the putative presence of cryptic species that, although potentially common, are typically not addressed in coral reef studies42. Nevertheless, it also demonstrates what information can be acquired with respect to genetic divergence patterns even in the case of non-ideal sampling datasets (our dataset may comprise a ‘typical’ coral collection dataset in this regard). In the context of conservation strategies, the disparity between morphology- and genetics-based species resolution highlights the necessity for robust molecular characterization to be able to resolve species diversity.
The three genera demonstrated very different degrees of genetic diversity and connectivity despite being sampled across the same environmental range and sites. This has implications for informing conservation efforts. Our genetic analyses suggest that Pocillopora is characterized by a higher level of within-species connectivity than Porites. A correlation between genetic dissimilarity and geographic distance as well as within-species genetic structuring (inferred by sNMF genetic clustering; Figs. 1 and 3) was only detected in Porites in line with less resolutive past studies43,44, and residuals (after TreeMix consensus tree generation) gave a considerably stronger within-species migration signal in Pocillopora than in Porites (Fig. 5). Maintenance of connectivity to ensure exchange of alleles is a key aim of resilience-based conservation management strategy8 and has been demonstrated to facilitate robustness and recovery to stress in reef ecosystems45,46,47. Therefore, while the higher number of species resolved in Pocillopora may suggest a greater conservation effort is required (greater species diversity to protect), its higher connectivity, in concert with its relatively high levels of species sympatry (in certain islands all species are present when considering only two sites, e.g. I05 Ducie or I08 Aitukaki, see also31), mean that core levels of diversity protection and resilience maintenance may be accomplished with a relatively focused effort. However, whether observed species are truly adapted to a given location or whether their presence is the result of/dependent on migration from a seed site (see below) must be carefully considered. In contrast to Pocillopora, the lower levels of connectivity and relatively higher levels of genetic structure in Porites suggest a greater spatial area of protection would be required to conserve genetic diversity and promote resilience16, especially considering the geographic isolation of P. evermanni (SSH1) in the eastern Pacific (I01-I03; Las Perlas, Coiba, and Malpelo) and the putative presence of yet another different genetic stock within this species in the Central Pacific32,43,44. While Pocillopora and Porites resolved into multiple distinct species, Millepora was resolved as a single species with highly endemic populations containing varying levels of admixture. In the context of maintaining genetic diversity, the Millepora population of I10 (Samoa) would be prioritized for protection over others, as this population has the highest proportion of admixture encompassing all three of the predicted ancestral populations. A recent study16 also detected distinct differences in connectivity between two reef-building species of the family Pocilloporidae that may, at least partially, be due to different reproductive modes (broadcast spawner, high connectivity, low genetic structure; brooder, low connectivity, high genetic structure). The reproductive mode could explain the differences in connectivity observed here but has not been characterized for the corals sampled. Further, generalizations based on other members of the respective genera are difficult, as Pocillopora and Porites both contain species that broadcast spawn and internally fertilize (brood), with some species exhibiting location-dependent reproductive modes48,49,50,51,52,53. Independent of identifying a causal factor for differences in connectivity, it is clear that species- and even population-specific characterizations of connectivity are a necessity in designing effective strategies (and conservation areas/zones) for genetic diversity maintenance.
Our dataset comprising samples of multiple species of Pocillopora and Porites across the same basin-wide range has enabled us to demonstrate contrasting evolutionary histories in these two genera, despite exposure to the same past thermal, and arguably, environmental regimes. The comparison of the genetic structures obtained in the two genera from samples coming from the same sites, along with the high sequencing depth (which led to a large number of genome-wide SNPs) on which the analyses were conducted, allows to at least partially offset the low local sample sizes for some SSHs. We found that although species geographic distribution for both Pocillopora and Porites are linked to past temperature variations, the underlying mechanisms driving this distribution vastly differ. Genetic distance was correlated to past thermal distance for Porites but not for Pocillopora (Supplementary Table 5). Given the significant correlation to thermal distance in Porites, one might have expected past thermal variation to play a role in Porites speciation/species divergence. However, the relative lack of divergent selection among Porites species suggests that these temperature variations did not have a role in species divergence and/or species isolation in this genus, in the sense that the elicited adaptive responses (resulting in at least some of the temperature outlier SNPs revealed by the genotype/environment association analyses) did not contribute to species differentiation. Rather, it would appear that the significant correlation to temperature distance is the product of correlation between the geographic and temperature distances (r = 0.564, P = 0.000, Supplementary Table 5) with the significant genetic and geographic correlation driven by the relatively low dispersal of Porites43,44,54. In Pocillopora, the lack of correlation between genetic distance and thermal distance is likely a consequence of the lack of correlation between genetic and geographic distances (Supplementary Table 5), diagnostic of high levels of dispersion and subsequent migration. Migration would enable a continuous influx of individuals with minimal genetic differentiation from seed sites to destination sites. These individuals would be adapted to the seed sites and may not survive thermal events such as cold spells linked to La Niña events at the sampled sites55. This combined high dispersal and sensitivity to environmental stress would concurrently explain the observed high overall species sympatry, lack of genetic distance correlations, yet the considerable effect of past temperature variation on the maintenance of species divergence (if not on the speciation process itself) in Pocillopora56.
The larger effect of past temperature variation on the evolutionary histories of the Pocillopora species could be symptomatic of a specialization/niche adaptation strategy (as opposed to being a more environment generalist like Porites)57 whereby any perturbation from the niche environment would necessitate an adaptive response due to a limited acclimation potential58. This explanation would seem less parsimonious given the acknowledged high morphological plasticity of the genus considered to be a driving factor in its evolutionary success37. However, a detailed comparison of temperature sensitivity among the genetically defined Pocillopora species is still lacking. It could also be argued that the adaptive potential of Porites is limited, but this is unlikely given the globally recognized resilience of this genus58,59,60. The genomic data produced here can provide the starting point for future functional analyses investigating the genes and pathways associated with the temperature outlier and GID SNPs identified, thus enabling cellular insights into adaptation potential and its underlying mechanisms.
Contemporary resilience-based conservation strategies do not fully consider how corals may adapt to future challenges5. Given the differences between species found across the same distribution here, it is interesting to speculate on species being more or less successful under climate change. Pocillopora species distribution appears more linked to the environment than Porites. Therefore, the Pocillopora genus likely displays a narrower set of physiological tolerances55,56,61. However, Pocillopora is known to be a speciose genus19, and while some of the extant species may be lost with the continuing and growing climatic perturbations predicted in the coming decades, surviving genotypes will have been selected for the future environments62. Coupled with the high dispersion capability of the genus—granting access to the widest possible range of habitats—this may result in a relatively successful strategy for the Pocillopora genus, enabling recolonization of future reefs by its more stress-tolerant species. Conservation efforts applied to such speciose, niche-adapted, and high-dispersal coral genera may catalyze the recovery of reefs by identifying those genotypes most likely to survive future perturbations, and where possible, protecting them from further non-climatic perturbations, while facilitating their already high-dispersal capacities to ensure the seeding of as wide a range of reef ecosystems as possible11. In contrast, generalist corals such as the Porites species identified in this study may be successful in the long term as long as the magnitude of perturbations are within the buffering capacity of their wider physiological tolerances57. However, as these buffering capacities are surpassed, the lower dispersal capacity of these species will mean a reduced ability to recolonize alternative environments and such genera may suffer losses5. In contrast to the targeted strategy of the niche-adapted corals, conservation of generalist corals should likely focus on mitigating local aggravating stressors (e.g., managing pollutant and nutrient input, limiting physical disruption due to coastal development, or preventing overfishing) to maximize the buffering capacity of the corals across as many of the available genotypes as possible.
Methods
Sample collection and environmental metadata acquisition
In the present study, we used corals which were sampled at 11 islands (Islas de las Perlas, Coiba, Malpelo, Rapa Nui, Ducie, Gambier, Moorea, Aitutaki, Niue, Upolu, and Guam) encompassing 32 sites (~3 sites per island) across an 18,000 km longitudinal transect from July 2016 to January 2017 during the first year of the Tara Pacific Expedition26. Coordinates and associated depth of the sampled sites and the number of samples collected are detailed in Supplementary Table 1. The mean depth at which the samples were collected was 9.3 ± 3.9 m (interquartile: 6.2–12 m). Three target species were sampled based on coral colony morphology: Pocillopora meandrina, Porites lobata, and Millepora cf. platyphylla. In addition, at Coiba (I02S01), three colonies of Millepora intricata33, that displayed a distinct branching morphology, were collected. Where possible, at each site, a minimum of three coral colonies were sampled and photographed as detailed in ref. 36 for a total of 106, 109, and 57 colonies of P. meandrina, P. lobata, and Millepora spp., respectively (Supplementary Table 1). Coral fragments were preserved in 15 ml of Lysing Matrix A beads (MP Biomedicals, Santa Ana, CA, USA) with 10 ml of DNA/RNA Shield buffer (Zymo Research, Irvine, CA, USA) and stored at −20 °C onboard the Tara vessel. The sampling, handling, nucleic acid extractions, and sequencing of coral colonies used in this study, along with the collection of their associated contextual metadata, are shortly described here, but their full descriptions can be found in two Tara Pacific data publications27,36.
Past sea surface temperature (SST) variations (such as, among other descriptors, the frequency, length, and intensity of hot and cold anomalies, etc.) supposedly had an impact on colony survival and recruitment, and therefore, structure the current genetic diversity in these species63. Therefore, at each sampling site, ocean skin temperature (11 and 12 μm spectral bands longwave algorithm) was extracted from 1 km resolution level-2 MODIS-Aqua and MODIS-Terra from 2002 to the sampling date and from level-2 VIIRS-SNPP from 2012 to the sampling date. Day and night overpasses were used to maximize data recovery. Following recommendations from NASA Ocean Color (OB.DAAC), only SST products of quality 0 and 1 were used. The nine closest pixels to the sampling sites were extracted. All the extracted pixels from the 3 platforms were then averaged daily to obtain daily SST averages and standard deviation time series for each sampling site, from 2002 to the sampling date. Different measures of the variation of these SSTs have been computed from these averages (as detailed in ref. 36) and used in the present study for the genotype/environment association and Gradient Forest analyses (see below).
DNA/RNA isolation, library preparation, and sequencing
For the Pocillopora, Porites, and Millepora colonies, coral fragments were disrupted using a high-speed homogenizer FastPrep-24 5 G Instrument (MP Biomedicals, Santa Ana, CA, USA). For DNA isolation, a preliminary enzymatic digestion of 1 h at 37 °C was applied on 500 µl of coral tissue with 50 µl of lysozyme (50 µl 10 mg/ml), 3 µl of mutanolysine (50 KU/ml), and 3 µl of lysostaphine (4 KU/ml). DNA and RNA were isolated with the Quick-DNA/RNA Kit (Zymo Research, Irvine, CA, USA). DNA and RNA were quantified on a Qubit 2.0 fluorometer with Qubit dsDNA BR (Broad range) and HS (High sensitivity) Assays and Qubit RNA HS Assay (ThermoFisher Scientific, Waltham, MA, USA), respectively.
For DNA, library preparation protocols were constructed to generate narrow-sized libraries around 300–800 bp using the NEBNext DNA Modules Products or the NEBNext Ultra II DNA Library prep kit (New England Biolabs, MA, USA). For Millepora, RNA libraries were prepared following the TruSeq Stranded mRNA sample preparation protocol. Extracted RNA (1 µg) was subjected to polyA+ selection using oligo(dT) beads, then converted into single-stranded cDNA using random hexamer priming. The second strand was generated to create double-stranded cDNA, purified by 1.8× AMPure XP bead cleanups, and ligated to TruSeq RNA barcoded adapters (Illumina, San Diego, CA, USA) or NEXTflex DNA barcoded adapters (Bioo Scientific, Austin, TX, USA). DNA and RNA libraries were sequenced using 151 bp paired-end read chemistry on a NovaSeq or HiSeq4000 Illumina sequencer (Illumina, San Diego, CA, USA) with a targeted yield of 108 reads per sample.
For both DNA and RNA, we removed short (<30 bp length) reads, low-quality nucleotides (Q score <20), adapter/primer sequences, and read pairs that mapped to the Enterobacteria phage PhiX174 genome (GenBank: NC_001422.1). For RNA, read pairs that mapped to ribosomal sequences were removed using SortMeRNA v2.164.
The raw sequences used for this study are deposited at the European Nucleotide Archive (ENA) at the EMBL European Bioinformatics Institute (EMBL-EBI) under the Tara Pacific Umbrella BioProject PRJEB47249. Additional methodologies for DNA isolation and sequencing are available in ref. 27.
SNP calling and filtering
For Pocillopora and Porites, we identified a set of genome-wide single-nucleotide polymorphisms (SNPs) by mapping the metagenomic reads to the Pocillopora cf. effusa and Porites lobata genomic references generated as part of the Tara Pacific expedition65 using the Genome Analysis Toolkit software (GATK, v3.7.0)66. We followed a modified version of the best practices protocol for variant discovery with GATK with manual filtering of the resulting variants. The following protocol was carried out for each species independently.
We aligned Illumina-generated 150-bp paired-end metagenomic reads sequenced from each colony to the predicted coding sequences of the Pocillopora cf. effusa and Porites lobata coral host reference genomes using Burrows–Wheeler Transform Aligner (BWA-mem, v0.7.15) with the default settings67. A read was considered a host contig if its sequence aligned with ≥95% sequence identity and with ≥50% of the sequence aligned. Host-mapped reads were sorted and filtered to remove sequences which contained >75% of low-complexity bases and <30% high-complexity bases using SAMtools v1.10.268 with the resultant bam files visualized using the Integrated Genomics Viewer69. The reference genomes were indexed with picardtools v2.6.0 (https://broadinstitute.github.io/picard/) using CreateSequenceDictionary before performing local realignment around small insertions and deletions (indels) using GATK’s RealignerTargetCreator and IndelRealigner to reduce false-positive variant identification and represent indels more parsimoniously.
After sample pre-processing, we called DNA variants (SNPs and indels) individually for each colony (GATK, HaplotypeCaller), generating one Genomic Variant Call Format (GVCF) file per sample. We then combined all per-colony GVCF files for a given island (ca. 9 files per island) into a single, multi-sample GVCF file (GATK, CombineGVCFs). This resulted in the generation of 11, island-specific combined GVCF files for each species. We then consolidated these 11 multi-sample GVCFs into a GenomicsDB database which allowed for subsequent variant calling across all island cohorts through a joint genotyping analysis (GATK, GenotypeGVCFs). Because the GenotypeGVCFs tool is capable of handling any ploidy level (or a mix of ploidies) intelligently, we did not specify the ploidy level in our function call. This analysis resulted in the generation of a single, “raw” Variant Call Format (VCF) file for each species in which all colonies were jointly genotyped. Because well-curated filtering resources necessary for GATK’s Variant Quality Score Recalibration (VQSR) tool were not available for either coral species, we filtered the raw VCF files manually using VCFtools v0.1.12 70 keeping only biallelic sites with minor allele frequencies ≥0.05, site-quality scores ≥30, and no missing data across colonies. This produced the “linked” datasets (5,937,714 and 1,971,638 variant sites with minimum read depth coverages ≥16 and 17 for Pocillopora and Porites, respectively) from which the SNPs were used for the identification of genomic islands of differentiation and the genomic signatures of selection. In order to avoid variant site linkage effects in downstream genetic analyses, we additionally filtered variants for linkage disequilibrium. We used the “prune” add-on feature in BCFtools v1.1171,72 to discard variants with a linkage disequilibrium (r2) ≥ 0.2 in a window of 1000 sites. This resulted in the “unlinked” datasets (347,243 and 183,222 variant sites for Pocillopora and Porites, respectively). The SNPs from this curated set were used to identify Secondary Species Hypotheses (SSHs) using individual-based phylogenies, genetic hierarchical clustering and species trees, and to test for population structure, admixture, and Genotype Environment associations, as described below.
For the detection of clonal lineages, we had to further reduce the size of these unlinked datasets to avoid unreasonable computing times. This was performed in Pocillopora by first further filtering on linkage disequilibrium (r2 ≥ 0.02) by BCFtools to a 27,382 variant site dataset for a first round of clone detection, and then by partitioning the individuals by SVDquartet lineages (see below). The coalescent testing of SSHs needed a further size reduction of the datasets, again on linkage disequilibrium, and keeping only one individual per putative genetic lineage per island (two replicates of 25 individuals and 636 variant sites and of 24 individuals and 1032 variant sites for Pocillopora and Porites, respectively).
Due to the absence of a reference genome at the time of SNP calling for Millepora cf. platyphylla, a ‘target gene’ approach was followed. Trimmed RNA-Seq metatranscriptomic sequences were subjected to de novo whole transcriptome assembly using Trinity73, after being further reduced to 23,755,336 reads by in silico normalization. The Trinity assembly produced 302,299 transcripts, which were then clustered into 40,560 unigenes (i.e., uniquely assembled transcripts) with an N50 of 1053 bp. Based on BUSCO, the assembled transcriptome was largely highly complete with 85.6% of the ortholog genes from the Eukaryota database being present with low fragmentation (3.6%), missing (10.8%), and duplication (16.7%) metrics. Within this transcriptome, we identified Millepora orthologs for an arbitrary set of stress and/or environment response genes from scleractinians and actinarians or genomic fragments from Pocillopora and Porites that contain temperature outlier SNPs obtained within the present study. The Millepora orthologs were obtained using QuickParanoid that takes a collection of files produced by InParanoid74 as input and finds ortholog clusters among species. The procedure used strictly followed the instructions published at http://pl.postech.ac.kr/QuickParanoid/. Briefly, for a given set of target genes, QuickParanoid first preprocesses each InParanoid output file by using BLASTall and then computes ortholog clusters. As for the other coral genera, metagenomic reads were aligned to this set of target genes using Burrows–Wheeler Transform Aligner (BWA-mem, v0.7.15) with the default settings67. Host-mapped reads were then sorted and processed using SAMtools v1.10.268 to generate respective bam files, and SNPs were identified as detailed above using GATK. The list of orthologs for the Millepora transcriptome contigs that contained SNPs is part of the resource files and scripts deposited in ref. 75. The SNP dataset was then filtered for biallelic status, SNPs only, quality, missingness, and minimum allele frequency with the same thresholds as for the other coral genera resulting in a final set of 446 variant sites. One individual with an excess of missing data (I09S03C026) was not included in the sNMF analysis. Filtering on linkage disequilibrium (r2 > 0.2) for further genetic analyses reduced this dataset to 243 unlinked variant sites in 56 individuals.
Species delimitation overview
Here we followed the integrative taxonomy approach14 by treating our morphology-guided sampling of the three coral species as primary species hypotheses (PSHs) from which we developed secondary species hypotheses (SSHs) through analysis of genetic diversity and colony macromorphology. We then formally tested the SSHs using coalescent analysis25.
Genetic delineation of species
To obtain an initial impression of the genetic diversity contained in the Pocillopora and Porites samples, we built an individual-based Maximum Likelihood phylogenetic tree using RAxML v8.2.1276. For this, the “unlinked SNPs” VCFs were translated to relaxed Phylip format using PGDSpider77, and raxmlHPC-PTHREADS was run on 40 cores, with a GTRCAT model and the number of bootstrap replicates determined by the autoFC option. The best ML tree with support value was then transferred in Newick format to MEGA X78 for graphical annotation.
A species tree was then built using SVDquartets79 as implemented in PAUP*80 v4.0a152 for all three genera. This non-Bayesian approach infers the relationship among quartets of taxa under a coalescent model and uses this information to build the species tree. Parallel computing on 50 threads was performed to analyze all possible quartets and 100 bootstrap replicates. The bootstrap consensus tree was then transferred to MegaX for graphical annotation.
To select which of the SVDquartet clades formed pertinent SSH, a hierarchical genetic clustering was also performed using the program sNMF81 with the snmf function from the LEA v2.8.082 R package. However, as this analysis is sensitive to clonality (B. Porro, pers. comm.), we first identified clonal lineages in the Pocillopora, Porites, and Millepora “unlinked” datasets using the R package Rclone v1.0.283 to identify the pairwise threshold distance between clonal individuals, and then used the mlg.filter function from the R package poppr v2.8.684,85 to assign individuals to clonal, or, more properly, multilocus lineages (MLL). We ran this clonal analysis on all samples for each of the 3 genera. In Pocillopora and Porites, due to a considerable lineage-correlating variation in pairwise distances, we also ran further analyses on SVD-grouping-defined subsets of samples. This analysis highlighted the presence of six MLL shared among 14 specimens in Pocillopora and five MLL shared among 12 specimens in Millepora. No shared MLL were found among Porites (Supplementary Table 3). The subsequent sNMF analyses were performed keeping only one ramet per clonal genet except for the 3 M. intricata samples that were identified as being a single MLL. sNMF determines the optimal number of ancestral populations from which the actual dataset could be issued through admixture. These analyses were performed on 50 cores and 10 repetitions for a number of possible ancestral populations that varied from 1 to 20, with an alpha parameter value of 10. The best number of ancestral clusters was determined by the entropy criterion, and a bar chart representing the individual admixture coefficient from each of these ancestral clusters was produced. To test for possible subclustering, a second round of sNMF analyses was performed separately on each of the ancestral clusters identified in the first round (with the same analysis parameters). As a further visualization of the genetic diversity within each of the 3 genera, we performed principal component analyses (PCA) based on individual genome-wide genotypes using the glPca function of the adegenet v1.7-15R package86,87. For Pocillopra and Porites, sNMF clustering and SVDquartet species-tree topology then guided the construction of secondary species hypotheses to be tested by coalescent analysis using Bayes factor delimitation with genomic data (BFD*)88,89. BFD analyses were not conducted in Millepora due to the low number of SNPs. The BFD* analyses were performed in BEAST2 2.6.290 following BFD* 2017 tutorial recommendations (https://beast2.blogs.auckland.ac.nz/tutorials/), with 48 path sampler sets of 500,000 MCMC repetitions, to sample in 10,000,000 MCMC iterations of SNAPP89 for Pocillopora and 750,000 MCMC repetitions and 1,000,000 MCMC iterations of SNAPP for Porites. These numbers of iterations were determined as minimal for convergence by first running standard SNAPP analyses in Beast2. Following the BFD* tutorial recommendations, the priors for the runs were computed from the tree height, for the estimation of Yule model birth rate, and the mean within-species divergence, as theta estimator, from the RAxML trees, with values of respectively 0.151 and 0.115 for Pocillopora and 0.156 and 0.107 for Porites, respectively. To select for the most pertinent SSH, the BFD* likelihood ranking of the SSH was performed twice in each genus on a duplicate set of individuals for the same variant sites.
Morphology analysis
To investigate the degree to which morphology may support or confound the genetics-based SSHs, we categorized the samples into morphotypes through morphological analysis and contrasted these groupings with the genetic resolutions. The M. intricata samples were excluded from the analysis. Photographs of entire colonies were taken for all samples using an underwater camera with 20 × 20 cm quadrats as a scale (photographs available online at https://store.pangaea.de/Projects/TARA-PACIFIC/Images/). Collection of morphological parameters was conducted using ImageJ v1.50e91, the image annotation plugin objectj v1.05j (https://sils.fnwi.uva.nl/bcb/objectj/), and custom macros with the photos corrected for perspective using the “interactive perspective” plugin with the quadrat as a reference.
The following morphological parameters were collected. For Pocillopora: branch tips density (calculated from the number of tips divided by the projected surface of the whole colony), tip length polyline (a line connecting the two extremities of the top of a branch following the curvature of the tip; n = 3), tip length ratio (the ratio between a straight line connecting the extremities of the top of a branch and the tip length polyline to detect the degree of meander shape of the branch tip; n = 3), tip width (n = 3), and colony roundness (the difference between the maximum and minimum diameters, and branch spacing between a randomly selected center branch and six adjacent branches; n = 6). For Porites: colony diameter, projected surface, growth form (columnar, massive or encrusting), lobe density (computed by the number lobes divided by the area of a 20 × 20 cm quadrat drawn on the colony photo using the ImageJ rectangle tool), and presence of ridges (present or absent). For Millepora: growth form (encrusting, laminar, columnar, branching columnar), column or plate density (computed by the number of columns or plates within the 20 × 20 cm quadrat), length of columns or plates (the length of a straight line connecting the extremities of the top of a column/plate; n = 1–6, when present), and width of columns/plates (n = 1–6, when present).
Morphometric analysis of the morphological parameters was conducted in R V4.1.0 using the same approach for all three genera. Unsupervised Random Forest analysis (randomForest::randomforest) was used to generate a proximity matrix upon which hierarchical clustering was performed to identify morphotypes (stat::hclust, factoextra::fviz_dend) with a threshold set so that the number of clusters equated to the number of genetically derived SSHs (Pocillopora and Porites), or the six SVD groupings (Millepora). The Gini coefficient was tested to measure the contribution of morphological variables in the homogeneity of the nodes of the decision trees. The proximity matrix was used as input to generate a principal coordinate analysis ordination (PCoA; stats::cmdscale) to visualize variance in morphology across the samples in accordance with the genetic lineage assignments. In addition, PERMANOVA (pairwiseAdonis::pairwise.adonis2; 999 permutations; P-adjusted Bonferroni correction applied; https://github.com/pmartinezarbizu/pairwiseAdonis) was used to test whether variance in morphology could be significantly explained by the genetic delineations.
Identification of Pocillopora and Porites SSH taxonomies
To assign species names to our SSH, two representatives for each SVD were mapped on the 2068 Pocillopora target-capture reference sequences from ref. 28 using BWA67. Samples were then genotyped for the 1559 unlinked diagnostic SNPs generated therein. Genotypes with a minimum read depth (DP) of 12× and nonsignificant strand biases (SP < 13) were called and filtered with BCFtools v1.1171,72. Between 1489 and 1524 SNPs were retrieved per sample. The resulting VCF was then combined with the one used for species delimitation in ref. 28, and assignment tests were performed from K = 2 to K = 30 using the program sNMF81 with the snmf function from the LEA82 R package, as previously described. From that, SSH correspondence with genomic species hypotheses (GSH) from ref. 28, and thus current taxonomy, was retrieved. Taxonomy was further confirmed by analysis of mitochondrial open reading frame (mtORF) sequences extracted from the metagenomic reads of the sampled Pocillopora colonies. Host metagenomic reads were mapped against a curated collection of species-diagnostic mtORF profiles29,37 using the Burrows–Wheeler Transform Aligner (BWA-mem, v0.7.15) with default settings67. Mapped reads were then sorted and processed using SAMtools (v1.10.2) to generate respective bam files71. Mapped reads were further filtered to retain only sequences that aligned to a given mtORF reference with ≥95% of sequence identity and with ≥80% of the sequence aligned. For each colony, a consensus mtORF sequence was generated from the aligned reads using the SAMtools mpileup feature followed by multiple sequence alignment in MUSCLE (v3.8.1551). Finally, colony consensus sequences and reference mtORF sequences31 were aligned with MAFFT version 7.4992, and a maximum likelihood phylogeny was performed in MegaX version 10.1.593 with bootstrap replicates to produce an mtORF phylogeny. A similar approach was applied to Porites, by mapping the metagenomic reads of one individual per genetic subcluster within each SSH to the P. lobata ortholog sequences of three species-diagnostic single-copy nuclear loci from ref. 32 (MM100, MM32, and ATPaseB). The orthologs of these three loci were identified in the Tara Pacific Porites lobata reference genome65 by BLAST from the sequences of an Hawaiian P. evermanni from32. SNPs were called with SAMtools mpileup and allelic variants sequences were obtained for each of these loci for each sample using the BCFtools consensus function on the resulting VCF file. The resulting fasta files were then added to the sequences from ref. 32 and processed following the protocols detailed therein to produce a POFAD genotype network94. Without a doubt, thorough species naming is a critical part of evolutionary and conservation biology95,96. However, it is equally important to acknowledge the limitations of morphology-targeted sampling approaches: our sampling did not include all known taxonomic diversity within the Pocillopora and Porites genera, and we also missed a good number of species types locations. For this reason, we kept our taxonomic resolution to the level offered by genetic-based analyses of described species and abstained from hypothetical and possibly misguided cryptic lineage namings. Insufficient SNPs and absence of the relevant resources limited such an analysis for Millepora.
Among- and within-species genetic differentiation
To further resolve evolutionary history in the Pocillopora and Porites species designations, we investigated introgression and pairwise divergence for the SSHs. Following ref. 97, introgression among lineages was computed with TreeMix 1.1398. The unlinked SNP dataset VCF was converted to TreeMix format using vcftools70 and PLINK99. The individuals were regrouped in populations according to their SSH and islands of origin, and the optimal number of admixture events was estimated at this level. For this, we ran TreeMix 10 times for 0 to 10 events and estimated the optimal number of events using the optM R package100. We then ran TreeMix 100 times for this optimal number of admixture/introgression events to produce a consensus tree and bootstrap values using the BITE 1.2.0008 R package101. The residual covariance matrix was estimated for the optimal number of admixture/introgression events and the consensus tree using TreeMix. The occurrence of the admixture/introgression events were verified by computing the relevant f4 indices34 and correlation in genetic content was further quantified by calculating pairwise f2 distances using AdmixTools2 (admixtools 2.0 R package, https://uqrmaie1.github.io/admixtools/index.html) after converting the unlinked SNP dataset VCF to the BED format using vcftools70 and PLINK 1.90b6.2199.
To quantify and characterize genomic divergence between the SSHs within Pocillopora and Porites, we computed pairwise Weir and Cockerham102 FST along the genome (500 bp sliding window) with vcftools 0.1.1770.
Environment as a predictor of genetic lineage distribution
The algorithm gradientForest was used to assess whether temperature-related parameters and sampling depth could be predictors for the geographic distribution of the genetic lineage distribution found in Pocillopora and Porites. The analysis was conducted in R (v4.2.2) using the library gradientForest (v0.1-32)35. Abundance data of the lineage at each sites were normalized using the logarithm of the abundance plus the minimum abundance observed (log(y + c) where c is the minimum positive abundance y), according to ref. 38. Sea surface temperature-related variables, described in ref. 36 and the mean depth at each sampling site, calculated from individual depth data available in ref. 36 were used as predictors. The gradient forest model was computed using 300 trees, a maximum of level of split of 2 (calculated following38), a correlation threshold of 0.5, and 101 bins as recommended in ref. 38).
Correlation of genetic lineages with geographic distance and past thermal regime
To assess for isolation-by-distance and a possible effect of temperature regime on the evolutionary trajectories of the species, we conducted correlation analyses sensu103. For Pocillopora and Porites, samples were again grouped according to their SSH and islands of origin (i.e. SVD4_I01, SVD4_I02… etc.) while Millepora samples were grouped according to island of origin. Within each genus, pairwise group FST distances were generated using vcftools 0.1.1770 from the “unlinked” SNPs dataset and used to generate pairwise matrices of FST/(1-FST). These distances, representative of genetic dissimilarity, were then tested for correlation against distance matrices representing geographic and past temperature (environmental) distances using the mantel function from the vegan 2.5-6 R package104. Geographic distances were based on GPS coordinates of islands (estimated by an average of the site coordinates). Euclidean past temperature distances were generated from a multiparameter past temperature dataset collected for each of the 38 sampled sites as part of the Tara Pacific consortium (as detailed in ref. 36) and used in a site-wise manner after conducting PCA to reduce its dimensionality from 63 initial parameters to the 5 highest-scoring PCs that cumulatively accounted for >80% of the observed variance (using R package FactoMineR v2.4)105.
Effect of environment on SSH divergence
To assess the influence of the environment (using past temperature variations as a proxy) on divergence between the SSHs in Pocillopora and Porites, we first used the reduced dimension past temperature variation dataset (see above) to identify temperature outlier SNPs whose allelic frequencies were linked to past temperature variation using redundancy analysis (RDA) following the exact protocol of Capblancq and Forester106. For this, the unlinked SNP dataset had first to be converted to ped format using vcftools. From this RDA, we identified temperature outlier SNPs, i.e., those SNPs whose allelic frequency distributions among populations were best explained by the local past temperature variation, as the 100 SNPs with the associated lowest P values (respectively P < 2.15 × 10−4 in Pocillopora and P < 1.72 × 10−3 in Porites). We then defined a set of genomic islands of differentiation (GIDs) as the 1% of the 500 bp bins with the highest mean FST value after conducting a further pairwise sliding window FST analysis as detailed above but with samples grouped according to SSH for each of the genera. We compared the proportion of these temperature outlier SNPs that were found in GIDs (see above) between the two genera. Finally, we investigated the proportion of the temperature outlier SNPs, and in particular those located in GIDs, that were predicted to be under divergent selection using the program FLink v1.0107. FLink operates on a dataset of linked loci and is based on the F-model which measures differences in allele frequencies (FST) that are partitioned into locus- and population-specific components reflecting selection and drift, respectively. FLink structures samples in a hierarchical model with the level of “population” nested in “group” and “group” nested in “higher hierarchy”. We equated “group” to the SSH and “population” to island of collection for the samples, and generated allele counts from the “linked SNPs” dataset as input using ATLAS108. To reduce computational complexity, the target genome can be subdivided, with individual FLink analyses run across each subdivision107. However, each subdivision must have a suitable number of variant sites to enable effective inference of demographic parameters with 10,000 suggested as a minimum . As such, we first ran one instance of FLink for every contig in the Pocillopora and Porites genomes with at least 10,000 variants (subsampling using BCFtools v1.971) using 4 cores per instance and a total of 800,000 MCMC iterations (300,000 burnin). Analysis showed that the demographic parameters converged at similar values across contigs within each of the respective genomes. To identify loci under selection in the remaining contigs (i.e. those with <10,000 variants), we therefore ran one instance of FLink per contig with parameters fixed according to the converged demographic parameters of the previous runs. Contig-wise results were consolidated and loci under divergent selection at the higher hierarchy level (i.e., between lineages; output file Posterior_A.txt) were assessed by calculating a false discovery rate (FDR) from the posterior alpha distributions following the software documentation using a custom python script. The Nextflow109 pipelines used to run the analyses and the Python and R scripts used to process the results are available on GitHub (https://github.com/didillysquat/TARA_host_popgen_2022).
Data availability
Sequence data used in this study are accessible under the Tara Pacific Umbrella BioProject PRJEB47249. More specifically, metagenomic sequencing raw data can be directly accessed under PRJEB52368. A collection of resource files including raw and filtered vcf files have been deposited on Zenodo https://doi.org/10.5281/zenodo.718096675.
Code availability
The code/scripts used to conduct the analyses are detailed on Zenodo https://doi.org/10.5281/zenodo.718096675. The Nextflow pipelines used to run the analyses and the Python and R scripts used to process the results are available on GitHub (https://github.com/didillysquat/TARA_host_popgen_2022).
References
Fisher, R. et al. Species richness on coral reefs and the pursuit of convergent global estimates. Curr. Biol. 25, 500–505 (2015).
Smith, S. V. Coral-reef area and the contributions of reefs to processes and resources of the world’s oceans. Nature 273, 225–226 (1978).
Gattuso, J.-P. et al. OCEANOGRAPHY. Contrasting futures for ocean and society from different anthropogenic CO2 emissions scenarios. Science 349, aac4722 (2015).
Eddy, T. D. et al. Global decline in capacity of coral reefs to provide ecosystem services. One Earth 4, 1278–1285 (2021).
Kleypas, J. et al. Designing a blueprint for coral reef survival. Biol. Conserv. 257, 109107 (2021).
Voolstra, C. R., Peixoto, R. S. & Ferrier-Pagès, C. Mitigating the ecological collapse of coral reef ecosystems. EMBO Rep. 24, e56826 (2023).
Wilkinson, C. Status of Coral Reefs of the World: 2008 (Australian Institue of Marine Science, 2008).
Mcleod, E. et al. The future of resilience-based management in coral reef ecosystems. J. Environ. Manage. 233, 291–301 (2019).
Voolstra, C. R. et al. Extending the natural adaptive capacity of coral holobionts. Nat. Rev. Earth Environ. 2, 747–762 (2021).
Peixoto, R. S., & Voolstra, C. R. (2023). The baseline is already shifted: marine microbiome restoration and rehabilitation as essential tools to mitigate ecosystem decline. Frontiers in Marine Science, 10. https://doi.org/10.3389/fmars.2023.1218531.
Beger, M. et al. Evolving coral reef conservation with genetic information. Bull. Mar. Sci. 90, 159–185 (2014).
Almany, G. R. et al. Connectivity, biodiversity conservation and the design of marine reserve networks for coral reefs. Coral Reefs 28, 339–351 (2009).
Fontoura, L. et al. Protecting connectivity promotes successful biodiversity and fisheries conservation. Science 375, 336–340 (2022).
Pante, E. et al. Species are hypotheses: avoid connectivity assessments based on pillars of sand. Mol. Ecol. 24, 525–544 (2015).
Selkoe, K. A. et al. The DNA of coral reef biodiversity: predicting and protecting genetic diversity of reef assemblages. Proc. Biol. Sci. 283, 20160354 (2016).
Buitrago-López, C. et al. Disparate population and holobiont structure of pocilloporid corals across the Red Sea gradient demonstrate species-specific evolutionary trajectories. Mol. Ecol. 32, 2151–2173 (2023).
Flot, J.-F., Couloux, A. & Tillier, S. Haplowebs as a graphical tool for delimiting species: a revival of Doyle’s “field for recombination” approach and its application to the coral genus Pocillopora in Clipperton. BMC Evol. Biol. 10, 372 (2010).
Richards, Z. T., Berry, O. & van Oppen, M. J. H. Cryptic genetic divergence within threatened species of Acropora coral from the Indian and Pacific Oceans. Conserv. Genet. 17, 577–591 (2016).
Pinzón, J. H. et al. Blind to morphology: genetics identifies several widespread ecologically common species and few endemics among Indo-Pacific cauliflower corals (Pocillopora, Scleractinia). J. Biogeogr. 40, 1595–1608 (2013).
Pante, E. et al. Use of RAD sequencing for delimiting species. Heredity 114, 450–459 (2015).
Erickson, K. L., Pentico, A., Quattrini, A. M. & McFadden, C. S. New approaches to species delimitation and population structure of anthozoans: two case studies of octocorals using ultraconserved elements and exons. Mol. Ecol. Resour. 21, 78–92 (2021).
Cowman, P. F. et al. An enhanced target-enrichment bait set for Hexacorallia provides phylogenomic resolution of the staghorn corals (Acroporidae) and close relatives. Mol. Phylogenet. Evol. 153, 106944 (2020).
Grinblat, M. et al. Biogeography, reproductive biology and phylogenetic divergence within the Fungiidae (mushroom corals). Mol. Phylogenet. Evol. 164, 107265 (2021).
Ramírez-Portilla, C. et al. Solving the coral species delimitation conundrum. Syst. Biol. 71, 461–475 (2022).
Carstens, B. C., Pelletier, T. A., Reid, N. M. & Satler, J. D. How to fail at species delimitation. Mol. Ecol. 22, 4369–4383 (2013).
Planes, S. et al. The Tara Pacific expedition—a pan-ecosystemic approach of the “-omics” complexity of coral reef holobionts across the Pacific Ocean. PLoS Biol. 17, e3000483 (2019).
Belser, C. et al. Integrative omics framework for characterization of coral reef ecosystems from the Tara Pacific expedition. Sci Data 10, 326 (2023).
Oury, N., Noël, C., Mona, S., Aurelle, D. & Magalon, H. From genomics to integrative species delimitation? The case study of the Indo-Pacific Pocillopora corals. Mol. Phylogenet. Evol. 184, 107803 (2023).
Gélin, P., Postaire, B., Fauvelot, C. & Magalon, H. Reevaluating species number, distribution and endemism of the coral genus Pocillopora Lamarck, 1816 using species delimitation methods and microsatellites. Mol. Phylogenet. Evol. 109, 430–446 (2017).
Schmidt-Roach, S., Miller, K. J., Lundgren, P. & Andreakis, N. With eyes wide open: a revision of species within and closely related to the Pocillopora damicornis species complex (Scleractinia; Pocilloporidae) using morphology and genetics. Zool. J. Linn. Soc. 170, 1–33 (2014).
Johnston, E. C., Wyatt, A. S. J., Leichter, J. J. & Burgess, S. C. Niche differences in co-occurring cryptic coral species (Pocillopora spp.). Coral Reefs 41, 767–778 (2022).
Hellberg, M. E., Prada, C., Tan, M. H., Forsman, Z. H. & Baums, I. B. Getting a grip at the edge: recolonization and introgression in eastern Pacific Porites corals. J. Biogeogr. 43, 2147–2159 (2016).
Boissin, E., Leung, J. K. L., Denis, V., Bourmaud, C. A. F. & Gravier-Bonnet, N. Morpho-molecular delineation of structurally important reef species, the fire corals, Millepora spp., at Réunion Island, Southwestern Indian Ocean. Hydrobiologia 847, 1237–1255 (2020).
Reich, D., Thangaraj, K., Patterson, N., Price, A. L. & Singh, L. Reconstructing Indian population history. Nature 461, 489–494 (2009).
Ellis, N., Smith, S. J. & Pitcher, C. R. Gradient forests: calculating importance gradients on physical predictors. Ecology 93, 156–168 (2012).
Lombard, F. et al. Open science resources from the Tara Pacific expedition across coral reef and surface ocean ecosystems. Sci. Data 10, 324 (2023).
Johnston, E. C. et al. A genomic glance through the fog of plasticity and diversification in Pocillopora. Sci. Rep. 7, 5991 (2017).
Marti-Puig, P. et al. Extreme phenotypic polymorphism in the coral genus Pocillopora; micro-morphology corresponds to mitochondrial groups, while colony morphology does not. Bull. Mar. Sci. 90, 211–231 (2014).
Paz-García, D. A. et al. Morphological variation and different branch modularity across contrasting flow conditions in dominant Pocillopora reef-building corals. Oecologia 178, 207–218 (2015).
Todd, P. A. Morphological plasticity in scleractinian corals. Biol. Rev. Camb. Philos. Soc. 83, 315–337 (2008).
Voolstra, C. R. et al. Consensus guidelines for advancing coral holobiont genome and specimen voucher deposition. Front. Mar. Sci. 8, 1029 (2021).
Bongaerts, P. et al. Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr. Biol. 31, 2286–2298.e8 (2021).
Boulay, J. N., Hellberg, M. E., Cortés, J. & Baums, I. B. Unrecognized coral species diversity masks differences in functional ecology. Proc. Royal Soc. B: Biol. Sci. 281, 20131580 (2014).
Baums, I. B., Boulay, J. N., Polato, N. R. & Hellberg, M. E. No gene flow across the Eastern Pacific Barrier in the reef building coral Porites lobata. Mol. Ecol. 21, 5418–5433 (2012).
Palumbi, S. R. Population genetics, demographic connectivity, and the design of marine reserves. Ecol. Appl. 13, 146–158 (2003).
Van Oppen, M. J. H. & Gates, R. D. Conservation genetics and the resilience of reef-building corals. Mol. Ecol. 15, 3863–3883 (2006).
Colton, M. A. et al. Coral conservation in a warming world must harness evolutionary adaptation. Nat. Ecol. Evol. 6, 1405–1407 (2022).
Brown, N. P., Forsman, Z. H., Tisthammer, K. T. & Richmond, R. H. A resilient brooding coral in the broadcast spawning Porites lobata species complex: a new endemic, introduced species, mutant, or new adaptive potential? Coral Reefs 39, 809–818 (2020).
Richmond, R. & Hunter, C. Reproduction and recruitment of corals: comparisons among the Caribbean, the Tropical Pacific, and the Red Sea. Mar. Ecol. Prog. Ser. 60, 185–203 (1990).
Fadlallah, Y. H. Reproduction in the coral Pocillopora verrucosa on the reefs adjacent to the industrial city of Yanbu (Red Sea, Saudi Arabia). Proc. Fifth Int. Coral Reef Cong., Tahiti 4, 313–318 (1985).
Sier, C. J. S. & Olive, P. J. W. Reproduction and reproductive variability in the coral Pocillopora verrucosa from the Republic of Maldives. Mar. Biol. 118, 713–722 (1994).
Stimson, J. S. Mode and timing of reproduction in some common hermatypic corals of Hawaii and Enewetak. Mar. Biol. 48, 173–184 (1978).
Schmidt-Roach, S., Miller, K. J., Woolsey, E., Gerlach, G. & Baird, A. H. Broadcast spawning by Pocillopora species on the Great Barrier Reef. PLoS ONE 7, e50847 (2012).
Polato, N. R., Concepcion, G. T., Toonen, R. J. & Baums, I. B. Isolation by distance across the Hawaiian Archipelago in the reef-building coral Porites lobata. Mol. Ecol. 19, 4661–4677 (2010).
Rodríguez-Troncoso, A. P., Carpizo-Ituarte, E. & Cupul-Magaña, A. L. Differential response to cold and warm water conditions in Pocillopora colonies from the Central Mexican Pacific. J. Exp. Mar. Bio. Ecol. 391, 57–64 (2010).
Magalon, H., Adjeroud, M. & Veuille, M. Patterns of genetic variation do not correlate with geographical distance in the reef-building coral Pocillopora meandrina in the South Pacific. Mol. Ecol. 14, 1861–1868 (2005).
Shinzato, C. et al. Whole-genome sequencing highlights conservative genomic strategies of a stress-tolerant, long-lived Scleractinian coral, Porites australiensis Vaughan, 1918. Genome Biol. Evol. 13, evab270 (2021).
Ziegler, M., Roder, C. M., Büchel, C. & Voolstra, C. R. Limits to physiological plasticity of the coral Pocillopora verrucosa from the central Red Sea. Coral Reefs 33, 1115–1129 (2014).
Darling, E. S. et al. Evaluating life-history strategies of reef corals from species traits. Ecol. Lett. 15, 1378–1386 (2012).
Sawall, Y. et al. Extensive phenotypic plasticity of a Red Sea coral over a strong latitudinal temperature gradient suggests limited acclimatization potential to warming. Sci. Rep. 5, 8940 (2015).
D’Croz, L. & Maté, J. L. Experimental responses to elevated water temperature in genotypes of the reef coral Pocillopora damicornis from upwelling and non-upwelling environments in Panama. Coral Reefs 23, 473–483 (2004).
Bairos-Novak, K. R., Hoogenboom, M. O., van Oppen, M. J. H. & Connolly, S. R. Coral adaptation to climate change: meta-analysis reveals high heritability across multiple traits. Glob. Chang. Biol. 27, 5694–5710 (2021).
Sully, S., Burkepile, D. E., Donovan, M. K., Hodgson, G. & van Woesik, R. A global analysis of coral bleaching over the past two decades. Nat. Commun. 10, 1264 (2019).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Noel, B. et al. Pervasive tandem duplications and convergent evolution shape coral genomes. Genome Biol. 24, 123 (2023).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, Inc., 2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Sonnhammer, E. L. L. & Östlund, G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 43, D234–D239 (2015).
Hume, B. C. C. et al. Resources from: disparate patterns of genetic divergence in three widespread corals across a pan-pacific environmental gradient highlights species-specific adaptation trajectories. https://doi.org/10.5281/zenodo.7180966 (2022).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Lischer, H. E. L. & Excoffier, L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Chifman, J. & Kubatko, L. Quartet inference from SNP data under the coalescent model. Bioinformatics 30, 3317–3324 (2014).
Swofford, D. L., PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. (Sinauer Associates, Sunderland, Massachusetts, 2003).
Frichot, E., Mathieu, F., Trouillon, T., Bouchard, G. & François, O. Fast and efficient estimation of individual ancestry coefficients. Genetics 196, 973–983 (2014).
Frichot, E. & François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929 (2015).
Bailleul, D., Stoeckel, S. & Arnaud-Haond, S. RClone: a package to identify MultiLocus Clonal Lineages and handle clonal data sets in r. Methods Ecol. Evol. 7, 966–970 (2016).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281 (2014).
Kamvar, Z. N., Brooks, J. C. & Grünwald, N. J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 6, 208 (2015).
Jombart, T. & Ahmed, I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Grummer, J. A., Bryson, R. W. Jr & Reeder, T. W. Species delimitation using Bayes factors: simulations and application to the Sceloporus scalaris species group (Squamata: Phrynosomatidae). Syst. Biol. 63, 119–133 (2014).
Bryant, D., Bouckaert, R., Felsenstein, J., Rosenberg, N. A. & RoyChoudhury, A. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917–1932 (2012).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Joly, S. & Bruneau, A. Incorporating allelic variation for reconstructing the evolutionary history of organisms from multiple genes: an example from Rosa in North America. Syst. Biol. 55, 623–636 (2006).
Bonito, V. E., Baird, A. H., Bridge, T., Cowman, P. F. & Fenner, D. Types, topotypes and vouchers are the key to progress in coral taxonomy: comment on Wepfer et al. (2020). Mol. Phylogenet. Evol. 159, 107104 (2021).
LaJeunesse, T. C. et al. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 28, 2570–2580.e6 (2018).
Louis, M. et al. Selection on ancestral genetic variation fuels repeated ecotype formation in bottlenose dolphins. Sci. Adv. 7, eabg1245 (2021).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Fitak, R. R. OptM: estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 6 https://doi.org/10.1093/biomethods/bpab017 (2021).
Milanesi, M. et al. BITE: an R package for biodiversity analyses. Preprint at bioRxiv https://doi.org/10.1101/181610 (2017).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228 (1997).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.5-6. 2019. https://CRAN.Rproject.org/package=vegan (2020).
Lê, S., Josse, J. & Husson, F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25, 1–18 (2008).
Capblancq, T. & Forester, B. R. Redundancy analysis: a Swiss Army Knife for landscape genomics. Methods Ecol. Evol. 12, 2298–2309 (2021).
Galimberti, M. et al. Detecting selection from linked sites using an F-model. Genetics 216, 1205–1215 (2020).
Link, V. et al. ATLAS: analysis tools for low-depth and ancient samples. Preprint at bioRxiv https://doi.org/10.1101/105346 (2017).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Tara Pacific Consortium. Tara Pacific Expedition Participants. at https://doi.org/10.5281/ZENODO.3777760 (2020).
Spalding, M., Mark D. Spalding, M. A., Ravilious, C., Green, E. P. & United Nations Environment Programme. World Atlas of Coral Reefs (University of California Press, 2001).
UNEP-WCMC, WorldFish Centre, WRI, TNC. Global distribution of warm-water coral reefs, compiled from multiple sources (listed in “Coral_Source.mdb”), and including IMaRS-USF and IRD (2005), IMaRS-USF (2005) and Spalding et al. (2001). data.unepwcmc.org/datasets/13 (Cambridge (UK): UNEP World Conservation Monitoring Centre, 2010).
Hume, B. C. C. & Voolstra, C. R. didillysquat/reefMapMaker: v0.1.10. https://doi.org/10.5281/zenodo.6826381 (2022).
Acknowledgements
Special thanks to the Tara Ocean Foundation, the R/V Tara crew and the Tara Pacific Expedition Participants110. We are keen to thank the commitment of the people and the following institutions for their financial and scientific support that made Tara Pacific expedition possible: CNRS, PSL, CSM, EPHE, Genoscope/CEA, Inserm, Université Côte d’Azur, ANR, and the Tara Ocean Foundation’s teams, crew, Président Etienne Bourgois and its partners agnès b., UNESCO-IOC, the Veolia Environment Foundation, Région Bretagne, Billerudkorsnas, Amerisource Bergen Company, Lorient Agglomeration, Smilewave, Oceans by Disney, the Prince Albert II de Monaco Foundation, L’Oréal, Biotherm, France Collectivités, Fonds Français pour l’Environnement Mondial (FFEM), the Ministère des Affaires Européennes et Etrangères, the Museum National d’Histoire Naturelle,. Tara Pacific would not exist without the continuous support of the participating institutes. The authors also particularly thank Serge Planes, Denis Allemand and the Tara Pacific consortium. This is publication number 25 of the Tara Pacific Consortium. The acknowledgments to local people and authorities who made this study possible can be accessed in the associated Zenodo deposition75. We would like to further thank Hans-Joachim Ruscheweyh for lending us computing time at ETH for some of the BFD* replicate runs. Work from BP, PF, DF was supported by the French Government (National Research Agency, ANR) through the grant “Coralgene” ANR-17-CE02-0020 as well as the “Investments for the Future” programs LABEX SIGNALIFE ANR-11-LABX-0028 and IDEX UCAJedi ANR-15-IDEX-0001. Work from BCCH was supported by AFF funding from the University of Konstanz, Germany. We would like to thank the SequAna Core Facility at the University of Konstanz for providing computing time. Work from EJA was supported by the French government (National Research Agency, ANR) through the grant “France Génomique” ANR-10-INBS-09. Work from GM is supported by JST SPRING, Grant Number JPMJSP2124. EB was supported by a Marie Curie fellowship MC-CIG-618480 to study the biogeography of Indo-Pacific fire corals.
Author information
Authors and Affiliations
Contributions
B.C.C.H. and C.R.V. wrote the main manuscript text. B.C.C.H., E.J.A., G.M., B.P., D.F., E.B., S.A., D.Z., Q.C., N.O., and H.M. analyzed the data. C.M., G.B., G.I., S.R., and E.B. collected the data. B.B., E.B., C.V., E.D., M.F., D.F., P.F., P.E.G., E.G., F.L., S.P., S.R., M.B.S., S.S., O.T., R.T., R.V.T., C.R.V., P.W., D.Z, S.P., and D.A. coordinated the Tara Pacific project, including this study. B.C.C.H., C.R.V., and E.A. contributed equally to the manuscript and D.F. coordinated the present study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Voolstra, C.R., Hume, B.C.C., Armstrong, E.J. et al. Disparate genetic divergence patterns in three corals across a pan-Pacific environmental gradient highlight species-specific adaptation. npj biodivers 2, 15 (2023). https://doi.org/10.1038/s44185-023-00020-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44185-023-00020-8
This article is cited by
-
Integrating cryptic diversity into coral evolution, symbiosis and conservation
Nature Ecology & Evolution (2024)
-
Multi-omics determination of metabolome diversity in natural coral populations in the Pacific Ocean
Communications Earth & Environment (2023)
-
Different environmental response strategies in sympatric corals from Pacific Islands
Communications Earth & Environment (2023)
