
Abstract
With anticipated expansion of agricultural areas for food production and increasing intensity of pressures stemming from land-use, it is critical to better understand how species respond to land-use change. This is particularly true for microbial communities which provide key ecosystem functions and display fastest responses to environmental change. However, regional land-use effects on local environmental conditions are often neglected, and, hence, underestimated when investigating community responses. Here we show that the effects stemming from agricultural and forested land use are strongest reflected in water conductivity, pH and phosphorus concentration, shaping microbial communities and their assembly processes. Using a joint species distribution modelling framework with community data based on metabarcoding, we quantify the contribution of land-use types in determining local environmental variables and uncover the impact of both, land-use, and local environment, on microbial stream communities. We found that community assembly is closely linked to land-use type but that the local environment strongly mediates the effects of land-use, resulting in systematic variation of taxon responses to environmental conditions, depending on their domain (bacteria vs. eukaryote) and trophic mode (autotrophy vs. heterotrophy). Given that regional land-use type strongly shapes local environments, it is paramount to consider its key role in shaping local stream communities.
Similar content being viewed by others

Introduction
Changes in land use are among the strongest drivers affecting species communities, diversity, and the ecosystem services they provide [1, 2]. Land-use changes do not only alter terrestrial ecosystems but also influence adjacent freshwater biodiversity and ecosystem functions in lakes and streams within the drainage systems [3, 4]. Freshwater ecosystems are thus considered sentinels to the impact of altered landscape processes since they are governed by the resulting changes in nutrient loads and exports of organic matter of vicinal areas [5]. Freshwater environments are particularly affected by the conversion from forested areas to agricultural lands through higher deposition of nutrients, dissolved organic matter, suspended solids and pollutants that alter water quality [6, 7]. At today’s numbers, about 38% of the ice-free land surface is dedicated to agricultural activities [8], and we have—by the year of 2022—crossed the milestone of 8 billion living humans on Earth. With the projected population growth to 10 billion by 2050 [9] we should expect human pressure for food production to expand agricultural areas and increase land use intensity [10] further affecting freshwater community structure and ecosystem functioning.
Community composition and biodiversity are widely used as surrogates to assess aquatic ecosystem status and functioning [11,12,13]. This is because water chemistry typically varies greatly even at short timeframes especially in smaller streams due to hydrological changes. Among several taxa, microbial communities emerge as a good bioindicator of fast environmental changes, due to their short generation times, large population densities and wide regional occurrence. They play an important role for ecosystem functioning by decomposing organic matter, transforming nutrients, and providing the basal food resource for higher trophic levels such as macroinvertebrates [14,15,16]. Accounting for the community structure of microbes has proven beneficial for explaining specific ecosystem functions [17]. Thus, taking a community perspective when investigating the impacts of land-use change on aquatic ecosystems is paramount. Most studies that investigate microbial community responses to environmental pressures commonly rely on ordination or cluster analyses that fail to account for potential species interactions and evolutionary constraints of environmental responses. During the last decade, species distribution modelling has become a common and fundamental tool to better understand and predict environmental filtering of communities [18]. Joint species distribution models (JSDM) in particular provide novel insight on community wide, yet taxa-specific responses with the advantages of taxa not being modelled independently but with an underlying joint structure related to abiotic (i.e., environment) and biotic filtering (i.e., species co-occurrences) simultaneously. Previous research on microbial stream communities has addressed the impacts of local environmental conditions [19, 20] and land-use types [21,22,23], including influences on biodiversity and function [24, 25]. However, the dependencies between regional land use type as a proxy variable and small-scale environmental conditions shaping communities are often neglected, leading to underestimated impacts of land use change. Using novel community level statistical advances provided by JSDM frameworks offers better understanding on the combined effects stemming from local environmental and regional land use pressures, and their respective contribution to microbial community composition and assembly processes. This is especially true as land-use impacts and changes in local environmental conditions in streams vary at different temporal scales, with land-use changes occurring at much longer time scales than any change in local stream conditions [3]. Even past land use is shown to influence water chemistry [26, 27]. Therefore, quantifying the extent of land-use effects on local environmental conditions will improve the understanding of microbial community patterns emerging from the interplay between regional land-use and local environmental conditions.
The goal of this study is to quantify the contribution of land-use types (forested vs. agricultural) in determining local environmental variables (i.e., water chemistry variables) and to uncover the impact of both, land-use, and environment, on microbial stream community assembly. With metabarcoding, we leverage bacteria and eukaryote community data from 120 stream sites in Finland to understand the resulting microbial community composition stemming from these drivers. Here we take advantage of a hierarchical JSDM to disentangle and quantify the species- and domain-specific (bacteria and eukaryote) drivers shaping community composition, while simultaneously accounting for phylogenetic constraints in species responses to the environment, as well as their trophic mode, i.e., being autotrophic or heterotrophic. This enables us to shed light on potential domain and/or trophy-specific responses to environmental conditions and susceptibility of different energy pathways. We further investigate the resulting impact of environmental filtering on microbial communities, using a region of common community profile approach.
Material and methods
Field sampling
Samples were collected in June–August 2020. In total, 120 stream sites were sampled from independent catchments totalling 60 agriculture and 60 forested sites (Fig. 1). The agriculture sites had >30% agricultural areas in the catchment area and the forested sites had <10% anthropogenic land use (i.e., agricultural areas + artificial surfaces) in the catchment area. The land cover and land use information were derived from the European land cover and land use classification CORINE (Coordination of information on the environment) dataset [28, 29] In the field, ten cobble-sized stones were randomly selected from different parts of each stream site for biofilm sampling. Biofilms were collected by scraping the surfaces of these stones (25 cm2 per stone) using a sponge (ca. 2 cm × 2 cm × 2 cm) and the resulting suspension was combined into a composite sample. At a few sites, less than ten stones were available. In these cases, multiple samples were collected from some larger stones so that the sampled stone surface area was the same for all sites. At each site, new sampling equipment were used, and samples were collected into sterilized sample containers. The samples were then stored frozen (−20 °C) and freeze-dried prior to subsequent laboratory analyses.

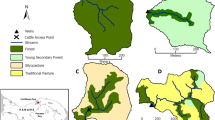
Map of (a) study area including sample region and sites, and (b–d) detailed sample sites with upper and lower panel including agricultural and forested sites, respectively, where colours represent the Region of Common Profile (RCP) based on community composition of (b) bacteria, (c) eukaryotes and (d) bacteria and eukaryotes.
Simultaneously with biofilm sampling, water temperature, pH, and conductivity were measured using a Hach HQ40d multimeter (Hach, Loveland, CO, USA) and water samples were collected. The water samples were analyzed in the laboratory for total phosphorus according to SFS-EN ISO 6878 and for total nitrogen according to standard SFS-EN ISO 11905–1 with a Hach Lange DR 5000 spectrometer (Hach Lange GmbH, Düsseldorf, Germany).
Environmental DNA and metabarcoding
DNA extraction, PCR and sequencing were performed at the Centre for Biodiversity Genomics, University of Guelph, Guelph, Canada. The lyophilized samples were sorted by weight into three categories (small, medium, and large). The volume of 1–6 mL of the Insect Lysis Buffer [30] with 10% Proteinase K was applied to the samples in proportion to their weight. The mixtures were vigorously vortexed, and 1 mL from each sample was subsampled into the Lysing matrix A, 2 mL tubes (MP Biomedicals) immediately. The lysates were ground in the TissueLyser at 30 Hz for 5 min, then incubated for 1 h at 56 °C, then frozen at −20° overnight, then incubated at 65 °C for 1 h 30 min. This procedure helped to break cellulous walls of the microscopic algae. DNA was extracted from the lysates with the modified automated DNA extraction protocol [31]. An additional precipitation on the Glass Fibre membrane with the consequent 5x dilution of DNA was used to obtain the best PCR result. Each sample was amplified in three replicas. The first round of PCR was performed with the following target-specific primers: Reuk454FWD1 (5'-CCAGCASCYGCGGTAATTCC-3') and V4r (5'-ACTTTCGTTCTTGAT-3') for eukaryotic 18 S rRNA region V4, ~380 base pairs; V8F (5'-ATAACAGGTCTGTGATGCCCT-3') and 1510 R (CCTTCYGCAGGTTCACCTAC) for eukaryotic 18 S rRNA regions V8-V9, ~330 base pairs; 341 F (5'-CCTACGGGNGGCWGCAG-3') and 805 R (5'-GACTACHVGGGTATCTAATCC-3') for bacterial 16 S rRNA regions V3-V4, ~460 base pairs. All primers were tailed with forward and reverse universal PacBio adaptors: PB1-forward (5'-GCAGTCGAACATGTAGCTGACTCAGGTCAC-3') and PB1-reverse (5'-TGGATCACTTGTGCAAGCATCACATCGTAG-3'). The following PCR1 conditions were used for 18 S V4 region: 94 °C for four minutes, followed by 35 cycles of 94 °C for 30 s, 46 °C for 30 s, 72 °C for 45 s, followed by 72 °C for ten minutes. The following PCR1 conditions were used for 18 S V8-V9 regions: 94 °C for four minutes, followed by 35 cycles of 94 °C for 30 s, 52 °C for 30 s, 72 °C for 45 s, followed by 72 °C for ten minutes. The following PCR1 conditions were used for 16 S V3-V4 regions: 94 °C for four minutes, followed by 35 cycles of 94 °C for 30 s, 50 °C for 30 s, 72 °C for 45 s, followed by 72 °C for ten minutes. PCR1 products were diluted with molecular grade ddH20 in 1:1 ratio. The 1 µL of diluted PCR1 product was used as a template in the second PCR (PCR2). The PCR2 was performed in 384-well microplate with the total volume of the PCR reaction 6 µL, using forward and reverse barcoded universal PacBio primers Plate-384v2 kit, and following PCR conditions: 94 °C for two minutes, followed by 20 cycles of 94 °C for 40 s, 64 °C for one minute, 72 °C for one minute, followed by 72 °C for ten minutes. The resulting asymmetrically labelled amplicon products were pooled for DNA sequencing and quantified with Qubit 2.0 Fluorometer (ThermoFisher). SMRTbell libraries for each fragment were prepared using Express Template Prep Kit 2.0 with barcoded adapters bc1015_BAK8B_OA, bc1016_BAK8B_OA, bc1017_BAK8B_OA. Library QC was performed using Agilent 100 Bioanalyzer. Sequencing was performed using Sequel platform (Pacific Biosciences) on a single 1 M SMRTcell v3 with eight-hour movie time and 20pM on-plate loading concentration. The resulting polymerase reads were demultiplexed in SMRTLink v.8 to generate datasets for each amplicon fragment. By-strand circular consensus reads (CCS) were generated for each dataset with minimum predicted accuracy 99%. Final CCS reads were further demultiplexed for individual samples based on exact scoring of forward and reverse barcoded primers. Subsequent bioinformatic analysis involved removal of loci-specific primer sequences with cutadapt [32] and assigned the sequences into operational taxonomic groups using vsearch [33] with 97% similarity for both bacteria and eukaryotes. Taxonomic assignment of representative sequences for each OTU was done with the classify.seqs command at Mothur [34] against Silva 138 [35] for bacteria and pr2 version 4.14.0 [36].
With high number of singleton OTUs we used species level annotation for bacteria and order level for the analysis of eukaryotes. The bacterial taxa with domain unknown or the ones classified as Chloroplast were removed from the further analysis as were the eukaryal taxa with domain unknown or phyla/supergroup level annotated as Eukaryota_unclassified. We further joint taxa with same unknown classification level for either genus or order. Taxa that were present in more than 10 of the analyzed 120 samples were kept for the statistical analysis which were conducted with the presence-absence data. For phylogenomic analysis we constructed Newick-formatted phylogenetic trees with MEGAX [37] using MUSCLE nt alignment [38] and UPGMA clustering.
Region of common community profile
We evaluated differences in community compositions and linked these to land use types by using a region of common profile (RCP) approach [39]. This was done by grouping sites based on their biological content, i.e., species occurrence data. Hence, communities within the same RCP are more similar in species composition than communities belonging to a different RCP. We used the dist.binary function from the ade4 package v. 1.7-16 [40] in the R environment [41] to compute the Jaccard dissimilarity matrix for binary data using the raw presence-absence data including bacteria and eukaryote OTUs. Subsequently we used the cascadeKM function, a K-means partitioning using a range of values of K, here between 1 and 10, from the package vegan v. 2.5-6 [42] to find the optimal number of clusters emerging from the community data using 1000 iterations. For optimal cluster evaluation, we used the well-established “calinski” criterion [43], included as output in the cascadeKM function. We used the resulting optimal data partition as RCPs, each reflecting sites of similar community composition.
Joint species distribution modelling
Emerging advances in statistical community modelling frameworks now allow us to specifically quantify how species’ responses to environmental variation depend on traits and their phylogenetic relationships [44]. We used a hierarchical joint species distribution modelling (JSDM) approach, the Hierarchical Modelling of Species Communities (HMSC; [44,45,46]). HMSC can incorporate trait and phylogenetic data of communities to improve estimations of species’ responses to environmental covariates while also quantifying their contribution to the explained variation. First, to understand how land-use types and the spatial location of site within land use type influenced the local environmental variables, we build an environmental model menv, where we included water conductivity, pH, temperature [°C], total phosphorus concentration [ug/l], and the total nitrogen to phosphorus ratio as response variables with the explanatory variables being land-use type (agricultural vs. forested) as a fixed effect and the spatial location of sites, given as latitudinal and longitudinal coordinates, as the random effect. This enabled us to construct a causal flow diagram quantifying the explained variation of environmental covariates related to the spatial setting and land-use type. Prior to the analysis we applied a log-transformation to the response variables water conductivity; phosphorus concentration and the N:P ratio; to better approximate normal distributions.
Subsequently, we constructed three different community models based on, (1) the bacterial community mbac, (2) the eukaryotic community meuk, and (3) the joint community mjoint including both domains, bacteria, and eukaryotes. We modelled spatially explicit eukaryotic and bacterial occurrences probabilities, based on OTUs, and their inference with environmental parameters. Our response variables were 217 OTUs, including 58 bacteria and 159 eukaryote taxa. The first two models, mbac and meuk, allowed us to quantify domain specific environmental drivers and the degree to which occurrence probabilities of included taxa depend on evolutionary constraints based on their phylogenetic relationship. With mjoint we modelled cross-domain species occurrences, specifically accounting for trophic modes within the community and investigated common community profiles in relation to land-use types. Each model was fitted with the same environmental variables, and the same spatially explicit structure. We included land-use, water conductivity, pH, temperature, total phosphorus concentration, and the total nitrogen to phosphorus ratio (N:P) as fixed effects. To account for the spatial structure of the study design, we included a spatially explicit random effect at the level of site, represented by latitudinal and longitudinal coordinates. Prior to the community model analyses, phosphorus concentrations as well as the N:P ratio were log-transformed.
HMSC incorporates a hierarchical layer accounting for phylogenetic constraints and traits in species responses to environmental variables [47]. In mbac and meuk we included the phylogenetic structure of the community as well as the categorical trait trophy accounting for autotrophic and heterotrophic species. We evaluated the strength of a potential phylogenetic signal to environmental variables within communities, i.e., if closely related species tend to have similar directional responses to environmental variables. The phylogenetic correlation parameter is denoted as rho, with possible values between 0 and 1 indicating whether the residual variance among the species is independent (0) or if their environmental responses are fully structured by their phenology (1). For mjoint we did not include phylogenetic information due to the cross-domain analysis but instead, in addition to the trait trophy, we also included the trait domain, enabling us to account for domain-specific trophic modes. We then use mjoint-specific parameters to quantify the domain- and trophy-specific environmental responses of taxa within the joint community.
We fitted all four above described models (menv, mbac, meuk and mjoint) with the Hierarchical Modelling of Species Communities (HMSC) R package Hmsc [46]. Since mbac, meuk and mjoint are based on binary presence absence data, the models followed a Bernoulli distribution with a probit link function, while menv followed a Gaussian distribution with an identity link function. We sampled the posterior distribution of each model with four Markov Chain Monte Carlo (MCMC) chains, each of which was run for 37,500 iterations, of which the first 12,500 were discarded as burn in. The chains had a thinning of 100 to yield 250 posterior samples per chain, resulting in 1000 posterior samples per model in total. We subsequently assessed MCMC convergence by examining the potential scale reduction factors [48] of the model parameters.
To examine the explanatory power of the models, we evaluated OTU-specific Tjur R2 [49] and AUC [50] values for the binary presence-absence models mbac, meuk and mjoint, and R2 values for menv. We quantified the drivers of community structure following Ovaskainen et al. [45] to partition explained variation among the fixed and random effects.
Results
Sequence data obtained
For bacteria a total of 378 (from 295–523) and for eukaryotes a total of 363 (from 85–865) PacBio Sequel sequence reads per sample were used in the analysis. The by-strand circular consensus reads (CCS) were generated for each dataset with a minimum predicted accuracy of 99%. With high quality due to the short read length and several cycles of sequencing, we used all reads in the analysis.
Land-use type structures community profiles at local environment
The data partitioning to highlight sites with common community profiles resulted in two optimal clusters for each domain, bacteria, and eukaryotes, as well as their joint composition (Fig. S1), namely RCP 1 and RCP 2 (Fig. 1b–d). We found strong separations between common community profiles depending on land-use type, where RCP 1 was strongly associated with agricultural, and RCP 2 with forested areas.
Local environmental conditions showed statistically significant differences between land-use types for all included environmental variables except temperature (Fig. 2). Linking the realized RCPs to the local environmental conditions at land-use types, we found that RCP 1 with high prevalence in agricultural areas was associated with higher values in water conductivity, pH, and phosphorus concertation compared to RCP 2 with high prevalence at forested sites (Fig. 2). The nitrogen to phosphorus ratio was on average higher in forested areas. Water temperature did not differ between land-use types.

Presence of joint bacteria and eukaryote community RCPs in relation to realized environmental conditions at land-use types.
Model convergence and fit
The MCMC convergence of all four models (menv, mbac, meuk and mjoint) was good, indicated by the potential scale reduction factor being ≪ 1.1 (Table 1). The models showed a satisfactory fit indicated by discriminatory and explanatory powers (Table 1).
Variance partitioning of land-use, local environment, and community relationships
We found that land-use type and sample location are strong predictors for local environmental conditions, as indicated by the respective R2 values of menv (Fig. 3). This is especially true for water conductivity and total phosphorus concentrations, where land-use and location together explained 98.5% and 94.7% of their total variation, respectively. The explanatory power of land-use type was particularly strong for conductivity (79%), pH (58%), and total phosphorus concentration (43%), while the explicit spatial effect contributed most to explained variation in nutrient concentrations, i.e., the nitrogen to phosphorus ratio (64%) as well as total phosphorus concentrations (52%) in streams. Only the variation of water temperature was independent of land-use type and location with <1% explained variation, respectively.

a Detailed causal flow diagram indicating explained variation (R2) of environmental variables by land-use type (top row left, forest vs. agriculture) and explicit spatial location (top row right). Thickness of arrows correspond to indicated R2 values. Middle row: symbols from left to right represent conductivity, water temperature, total phosphorus concentration, total nitrogen to phosphorus ratio, and pH with respective Tjur R2 values in explaining eukaryotic (meuk bottom row left) and bacterial (mbac bottom row right) species occurrences at the community level. Note that arrow thickness is proportional among Tjur R2 values (red and blue, meuk and mbac respectively) and among R2 values (green and yellow, menv). b Summarized causal flow diagram indicating mean explained variation of environmental variables by land-use type and location (R2 from menv, black), and fractions of explained variation of land-use and location as well as environmental variables on the joint community (Tjur R2 from mjoint, grey).
For both, eukaryote (meuk) and bacteria (mbac) communities, taxa occurrences were influenced similarly, in terms of proportions of the explained variation by the fixed and random effects (Fig. 3a). Water conductivity, phosphorus concentration, and pH were the strongest predictors, each alone exceeding the effect of land-use type as proxy variable as fixed effect. However, the spatial random effect had the second strongest explanatory power for species occurrences after pH, in both models, meuk and mbac.
When considering all eukaryotic and bacterial taxa together as joint community (mjoint), we found that of the total explained variation in species occurrences, about two thirds (61.9%) can be attributed to the local environmental conditions, while effects of the proxy variable land-use type (14.1%) and the explicit spatial location (24%) only accounted for one third of variation combined (Figs. 3b, S2). However, roughly 70% of the variation in local environmental variables can be attributed to land-use and location (Fig. 3b).
Species-environment responses depend on domain-specific trophic modes
Using the joint community model (mjoint) to account for domain-specific response types to environmental variables, we found that the effects of environmental conditions differed across bacteria and eukaryotes as well as across autotrophs and heterotrophs (Fig. 4). Among all autotrophic eukaryotes, individual taxa showed both positive and negative responses to the local and regional variables, except for total phosphorus concentrations, which generally increased the probability of taxon occurrence. Conversely, heterotrophic eukaryotes that showed statistically supported responses were mainly negatively associated with the included variables (Fig. 4a). While autotrophic bacteria were more likely to occur at higher pH values and in agricultural than in forested areas, the heterotrophic bacteria were less likely to occur in nutrient rich waters, being more favoured by warmer waters in forested areas (Fig. 4a). Noteworthy, there was a moderately strong phylogenetic signal in both bacteria (rho = 0.71, SD = 0.07) and eukaryote communities (rho = 0.75, SD = 0.08), suggesting that closely related species in each community tended to respond similarly to environmental variables (Fig. 4b).

a Density distribution of domain- and trophy- specific environment relationships with statistical support of 90% posterior probability, illustrated by standardized variable effects from mjoint. The number of statistically supported responses of taxa per panel group is indicated by n. b Phylogenetically structured directional responses of taxa occurrences to variables (left mbac, right meuk). Responses that are positive with at least 90% posterior probability are shown by red, responses that are negative with at least 90% posterior probability are shown in blue. Responses that did not gain strong statistical support are shown in white. Tree tip labels and taxonomic classifications are given in Supplementary Tables S1, S2 and Figs. S2, S3.
Discussion
Land-use types have a profound effect on freshwater communities, their biodiversity and on the ecosystem functions they provide [1, 51]. However, our results underline that regional land-use effects determine microbial community assembly by primarily shaping local environmental conditions. Here we demonstrated that (i) microbial stream community composition is closely linked to land-use type, (ii) local environmental conditions, driving microbial species responses, mediate the effects of land-use on microbial communities, and (iii) taxon responses show systematic variation to environmental variables, depending on their domain (bacteria vs. eukaryote) and trophic mode (autotrophy vs. heterotrophy).
We found a clear differentiation between the community compositions depending on land use type. This was true not only for the bacterial community but also for the eukaryotic community as well as for their joint composition. For each of the three community types two regions of common community profile (RCP) emerged, each reflecting similarity in community composition with affinities to either agricultural or forested areas, without any a priori assumptions regarding environmental conditions at the respective sites. This clear separation beyond taxonomic domains, suggests that environmental filtering and species sorting are the main processes underpinning the microbial community structure [52,53,54]. Different land use type-associated community composition also point to differences in water quality and microbial activity with altered stream metabolism possibly affecting higher trophic levels through altered food web structure [14, 51]. The strong observed impact of environmental filtering on the microbial meta-community supports their potential as bioindicators for the assessment of environmental conditions [55].
The effects of regional land-use on the local environmental conditions (e.g., water physico-chemistry) are often overlooked when investigating land use effects on communities [21, 56]. This may owe to the common aim to disentangle the respective contribution of land use, such as urbanized, agricultural, or forested areas, on species or communities in a certain environmental setting. However, here we show that regional land-use types overwhelmingly determine local key environmental drivers of microbial stream communities. While our results demonstrate that land use, when considered as large-scale proxy variable, explained less variation of species occurrences than local environmental conditions, the effects of land use on the variation of local stream conditions was substantial. This suggests that regional land use effects determine local stream conditions that in turn are the main driver of microbial stream community assembly. We argue that the full impact of land use may be underestimated when failing to consider that local environmental conditions mediate regional land use effects. The strongest environmental drivers explaining community composition, were also those which were best explained by land-use types, namely, water conductivity, water pH and phosphorus concertation, with statistically significant differences between agricultural and forested areas. However, we acknowledge that while our included explanatory covariates do reflect general parameters of local water quality, we did not consider any organic matter compositions here, that may have added a more detailed understanding in the mechanisms of community structures between land use types. Water pH-levels have been previously demonstrated to be a major driver of microbial stream communities [57, 58]. Our results support this finding with pH having the highest explanatory power of the fixed effects in all three community models, meuk, mbac and mjoint. We found high levels of pH values and water conductivity to be associated with agricultural land use. Microbial community responses to elevated values in both, pH and conductivity, have been linked to increased respiration rates, indicating stress responses, with the potential to destabilize microbial ecosystem function [51].
Although we found consistent responses of communities belonging to different domains, i.e., eukaryote vs. bacterial community, as well as the joint cross domain community, we found systematic variation of species responses to environmental variables and land-use type, depending on domain-specific trophic modes. Autotrophic eukaryotes show mainly positive associations to phosphorus concentration, while displaying a bimodal distribution of both negative and positive responses of included taxa to the remaining environmental variables, including land use type, suggesting that this group of species comprises members with a heterogeneous range of environmental niches. Heterotrophic eukaryotes on the other hand seemed more constraint in their niche space, showing mainly negative responses to realized environmental variables. While heterotrophic bacteria exhibited similar negative responses to nutrient concentrations as their eukaryotic counterparts, they had opposite responses to water temperature and land-use type, and no detected association with water conductivity. This indicates that heterotrophic bacteria and eukaryotes are associated with different environmental conditions, likely excluding competitive interactions as well as environment-specific energy flows. The fact that autotrophic bacteria only showed statistically supported responses to pH and land use points towards their robustness to environmental drivers and wide-ranging niche space. These trophy-specific responses are supported by the detected signal of phylogenetically structured environmental responses in both communities, signifying phylogenetic niche conservatism. When dividing responses of energy flow pathways into ‘green’, autotroph-based, and ‘brown’, heterotroph-based [59, 60], our results suggest that the brown pathways are more susceptible to environmental change and land use types than the green. Depending on the environmental setting as well as on primary consumer preference, shifts in such energy flow pathways may have ecosystem wide implications. Highlighting these different domain and trophy specific response types at the community level, and revealing their potential influence on ecosystem function, is not possible by applying the commonly used statistical tools such as ordination analyses, but requires the use of more timely advances in community ecology such as JSDM. Considering that land-use type is a strong determinant of the local environment, it will be important to not only consider its impact on species communities as a large-scale proxy variable, but also the even stronger local impact through mediated environmental conditions.
Our findings challenge the common approach to consider impacts of land-use change and local environmental variables on microbial communities independently from each other. We show that most of the explained variation in species responses is attributed to the local conditions, which are in fact to the largest part determined by regional land-use types. This strong impact of land-use on community assembly across domains redefines our current understanding towards the expanding pressures stemming from land-use change, provides better understanding to community wide responses, and may ultimately support stream ecosystem conservation efforts.
Data availability
Raw sequence and metadata can be found in the European Nucleotide Archive (ENA) under the project PRJEB62688.
References
Allan E, Manning P, Alt F, Binkenstein J, Blaser S, Blüthgen N, et al. Land use intensification alters ecosystem multifunctionality via loss of biodiversity and changes to functional composition. Ecol Lett. 2015;18:834–43.
Pereira HM, Navarro LM, Martins IS. Global biodiversity change: The Bad, the good, and the unknown. Annu Rev Environ Resour. 2012;37:25–50.
Soininen J, Bartels P, Heino J, Luoto M, Hillebrand H. Toward more integrated ecosystem research in aquatic and terrestrial environments. Bioscience. 2015;65:174–82.
Scherer-Lorenzen M, Gessner MO, Beisner BE, Messier C, Paquette A, Petermann JS, et al. Pathways for cross-boundary effects of biodiversity on ecosystem functioning. Trends Ecol Evol. 2022;37:454–67.
Williamson CE, Dodds W, Kratz TK, Palmer MA. Lakes and streams as sentinels of environmental change in terrestrial and atmospheric processes. Front Ecol Environ. 2008;6:247–54.
Wilson HF, Xenopoulos MA. Effects of agricultural land use on the composition of fluvial dissolved organic matter. Nat Geosci. 2009;2:37–41.
Scanlon BR, Jolly I, Sophocleous M, Zhang L. Global impacts of conversions from natural to agricultural ecosystems on water resources: Quantity versus quality. Water Resour Res. 2007;43:W03437.
FAO. World food and agriculture: statistical yearbook. 2021. Food and Agriculture Organization Rome.
United Nations Department of Economic and Social Affairs PD. World Population Prospects 2022: Summary of Results. UN DESA/POP/2022/TR/NO. 3. 2022.
Stehfest E, van Zeist WJ, Valin H, Havlik P, Popp A, Kyle P, et al. Key determinants of global land-use projections. Nat Commun. 2019;10:1–10.
Ector L, Rimet F Using bioindicators to assess rivers in Europe: An overview. Modelling Community Structure in Freshwater Ecosystems 2005; 7–19.
Burger J. Bioindicators: types, development, and use in ecological assessment and research. Environ Bioindic. 2006;1:22–39.
Lear G, Ancion PY, Harding J, Lewis GD. Use of bacterial communities to assess the ecological health of a recently restored stream. N Z J Mar Freshwater Res. 2012;46:291–301.
Findlay S. Stream microbial ecology. J North Am Benthol Soc. 2010;29:170–81.
Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive earth’s biogeochemical cycles. Science (1979). 2008;320:1034–9.
Battin TJ, Besemer K, Bengtsson MM, Romani AM, Packmann AI. The ecology and biogeochemistry of stream biofilms. Nat Rev Microbiol. 2016;14:251–63.
Graham EB, Knelman JE, Schindlbacher A, Siciliano S, Breulmann M, Yannarell A, et al. Microbes as engines of ecosystem function: When does community structure enhance predictions of ecosystem processes? Front Microbiol. 2016;7:1–10.
Warton DI, Blanchet FG, O’Hara RB, Ovaskainen O, Taskinen S, Walker SC, et al. So Many Variables: Joint Modeling in Community Ecology. Trends Ecol Evol. 2015;30:766–79.
Burdon FJ, Bai Y, Reyes M, Tamminen M, Staudacher P, Mangold S, et al. Stream microbial communities and ecosystem functioning show complex responses to multiple stressors in wastewater. Glob Chang Biol. 2020;26:6363–82.
Zeglin LH. Stream microbial diversity in response to environmental changes: Review and synthesis of existing research. Front Microbiol. 2015;6:1–15.
Laperriere SM, Hilderbrand RH, Keller SR, Trott R, Santoro AE. Headwater stream microbial diversity and function across agricultural and urban land use gradients. Appl Environ Microbiol. 2020;86:e00018-20.
Cooper SD, Lake PS, Sabater S, Melack JM, Sabo JL. The effects of land use changes on streams and rivers in mediterranean climates. Hydrobiologia. 2013;719:383–425.
Le HT, Rochelle-Newall E, Auda Y, Ribolzi O, Sengtaheuanghoung O, Thébault E, et al. Vicinal land use change strongly drives stream bacterial community in a tropical montane catchment. FEMS Microbiol Ecol. 2018;94:1–16.
Qu X, Ren Z, Zhang H, Zhang M, Zhang Y, Liu X, et al. Influences of anthropogenic land use on microbial community structure and functional potentials of stream benthic biofilms. Sci Rep. 2017;7:1–12.
Chavarria KA, Saltonstall K, Vinda J, Batista J, Lindmark M, Stallard RF, et al. Land use influences stream bacterial communities in lowland tropical watersheds. Sci Rep. 2021;11:21752.
Maloney KO, Weller DE. Anthropogenic disturbance and streams: Land use and land‐use change affect stream ecosystems via multiple pathways. Freshw Biol. 2011;56:611–26.
Maloney KO, Feminella JW, Mitchell RM, Miller SA, Mulholland PJ, Houser JN. Landuse legacies and small streams: identifying relationships between historical land use and contemporary stream conditions. J North Am Benthol Soc. 2008;27:280–94.
Finnish Environment Institute. VALUE - Valuma-alueen rajaustyökalu, http://paikkatieto.ymparisto.fi/value/. 2020.
Finnish Environment Institute. CORINE Land Cover 2012, https://www.syke.fi/avointieto.
Ivanova N, Grainger C, Hajibabaei M. Glass fiber DNA extraction: A new inexpensive method for high throughput DNA isolation. CCDB Advances, Methods Release 2006;3. https://ccdb.ca/site/wp-content/uploads/2016/09/CCDB_DNA_Extraction.pdf.
Ivanova N, Kuzmina M, Fazekas A CCDB Protocols: Manual protocol employing centrifugation: glass fiber plate DNA extraction protocol for plants, fungi, echinoderms and mollusks, http://ccdb.ca/site/wp-content/uploads/2016/09/CCDB_DNA_Extraction-Plants.pdf. 2011.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016;2016:1–22.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6.
Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L, et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013;41:597–604.
Stecher G, Tamura K, Kumar S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol Biol Evol. 2020;37:1237–9.
Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Foster SD, Givens GH, Dornan GJ, Dunstan PK, Darnell R. Modelling biological regions from multi-species and environmental data. Environmetrics. 2013;24:489–99.
Dray S, Dufour A-B. The ade4 Package: Implementing the Duality Diagram for Ecologists. J Stat Softw. 2007;22:1–20.
R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. www.r-project.org. 2020. Vienna, Austria.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package. R,. https://CRAN.R-project.org/package=vegan. 2019.
Caliński T, Harabasz J. A dendrite method for cluster analysis. Commun Stat-theory Methods. 1974;3:1–27.
Ovaskainen O, Abrego N. Joint species distribution modelling: with applications in R. 2020. Cambridge University Press, Cambridge.
Ovaskainen O, Tikhonov G, Norberg A, Guillaume Blanchet F, Duan L, Dunson D, et al. How to make more out of community data? A conceptual framework and its implementation as models and software. Ecol Lett. 2017;20:561–76.
Tikhonov G, Opedal ØH, Abrego N, Lehikoinen A, de Jonge MMJ, Oksanen J, et al. Joint species distribution modelling with the r-package Hmsc. Methods Ecol Evol. 2020;11:442–7.
Abrego N, Norberg A, Ovaskainen O. Measuring and predicting the influence of traits on the assembly processes of wood-inhabiting fungi. J Ecol. 2017;105:1070–81.
Gelman A, Rubin DB. Inference from iterative simulation using multiple sequences. Statistical Science. 1992;2:45–52.
Tjur T. Coefficients of determination in logistic regression models—A new proposal: The coefficient of discrimination. Am Stat. 2009;63:366–72.
Pearce J, Ferrier S. Evaluating the predictive performance of habitat models developed using logistic regression. Ecol Modell. 2000;133:225–45.
Dang C, Kellner E, Martin G, Freedman ZB, Hubbart J, Stephan K, et al. Land use intensification destabilizes stream microbial biodiversity and decreases metabolic efficiency. Sci Total Environ. 2021;767:21752.
Jiao S, Yang Y, Xu Y, Zhang J, Lu Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020;14:202–16.
Logue JB, Lindström ES. Species sorting affects bacterioplankton community composition as determined by 16S rDNA and 16S rRNA fingerprints. ISME J. 2010;4:729–38.
Van Der Gucht K, Cottenie K, Muylaert K, Vloemans N, Cousin S, Declerck S, et al. The power of species sorting: Local factors drive bacterial community composition over a wide range of spatial scales. Proc Natl Acad Sci USA. 2007;104:20404–9.
Simonin M, Voss KA, Hassett BA, Rocca JD, Wang SY, Bier RL, et al. In search of microbial indicator taxa: shifts in stream bacterial communities along an urbanization gradient. Environ Microbiol. 2019;21:3653–68.
Hosen JD, Febria CM, Crump BC, Palmer MA. Watershed urbanization linked to differences in stream bacterial community composition. Front Microbiol. 2017;8:1–17.
Fierer N, Morse JL, Berthrong ST, Bernhardt ES, Jackson RB. Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology. 2007;88:2162–73.
Liu S, Ren H, Shen L, Lou L, Tian G, Zheng P, et al. pH levels drive bacterial community structure in the Qiantang River as determined by 454 pyrosequencing. Front Microbiol. 2015;6:1–7.
Halvorson HM, Wyatt KH, Kuehn KA. Ecological significance of autotroph–heterotroph microbial interactions in freshwaters. Freshw Biol. 2020;65:1183–8.
Moore JC, Berlow EL, Coleman DC, De Suiter PC, Dong Q, Hastings A, et al. Detritus, trophic dynamics and biodiversity. Ecol Lett. 2004;7:584–600.
Acknowledgements
The study was funded by the Strategic Research Council of the Academy of Finland (Project 312650 BlueAdapt) and supported by the Kone Foundation grant 202102289 (to JS), the Academy of Finland grant 309581 (to OO), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme; grant agreement no. 856506; ERC-synergy project LIFEPLAN (to OO). Open access funded by Helsinki University Library.
Author information
Authors and Affiliations
Contributions
BW, CGR, JH, VP, AT, JS and OO jointly conceived the study. VP and AT designed the field sampling protocol, with input from JS, and collected samples. MK and EVK performed DNA extractions, PCR and sequencing. JH performed the bioinformatics analyses and species annotation. BW and CGR jointly performed the statistical analyses with contribution from OO. BW wrote the first draft of the manuscript with input from all authors. All authors edited and revised the manuscript resulting in the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weigel, B., Graco-Roza, C., Hultman, J. et al. Local eukaryotic and bacterial stream community assembly is shaped by regional land use effects. ISME COMMUN. 3, 65 (2023). https://doi.org/10.1038/s43705-023-00272-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43705-023-00272-2
