
Abstract
Multiomics has shown promise in noninvasive risk profiling and early detection of various common diseases. In the present study, in a prospective population-based cohort with ~18 years of e-health record follow-up, we investigated the incremental and combined value of genomic and gut metagenomic risk assessment compared with conventional risk factors for predicting incident coronary artery disease (CAD), type 2 diabetes (T2D), Alzheimer disease and prostate cancer. We found that polygenic risk scores (PRSs) improved prediction over conventional risk factors for all diseases. Gut microbiome scores improved predictive capacity over baseline age for CAD, T2D and prostate cancer. Integrated risk models of PRSs, gut microbiome scores and conventional risk factors achieved the highest predictive performance for all diseases studied compared with models based on conventional risk factors alone. The present study demonstrates that integrated PRSs and gut metagenomic risk models improve the predictive value over conventional risk factors for common chronic diseases.
Similar content being viewed by others

Main
Multiomic technologies have uncovered potential biomarkers for various common age-related diseases, including cardiovascular disease, diabetes, liver disease, dementia and cancer1,2,3,4,5,6. Although conventional risk prediction typically relies on demographic (for example, age or sex), anthropomorphic (for example, body mass index (BMI)), lifestyle factors and disease-specific clinical laboratory measurements (for example, blood pressure (BP), non-high-density lipoprotein (HDL)-cholesterol, mammographic density, creatinine, glycated hemoglobin (HbA1c)), the recent emergence of multiomics means that it is now possible to measure and integrate whole classes of biomolecular and cellular factors for the purposes of building multiomic risk scores.
PRSs, a quantitative measure of genetic predisposition for a phenotype, have demonstrated validity and potential clinical utility in risk prediction for various common diseases7,8,9,10, for example, in cardiovascular disease11,12,13,14, cancers15,16, diabetes mellitus17,18,19 and ankylosing spondylitis20. Given the potential of a genome-wide genotyping array as a one-time, relatively inexpensive assay from which hundreds of PRSs can be calculated, PRSs are being assessed in clinical studies for healthcare systems around the world9,11,21.
The gut microbiota (the collection of microorganisms inhabiting the human gastrointestinal tract) has also been shown to have a role in many common diseases22,23,24. Gut microbial signatures have been associated with mortality and incident diseases in the general population, such as type 2 diabetes (T2D) and liver and respiratory diseases4,25,26,27,28,29, suggesting the potential of the gut microbiome in disease risk prediction. Notably, although genome-wide association studies have revealed the human genetic basis of the gut microbiome30,31,32, it is apparent that the heritability of the gut microbiome is relatively low and cross-generational familial microbiome similarity is largely associated with cohabitation33,34,35.
Given that they are based on robust scalable technologies, use noninvasive sampling and have been applied in numerous disease risk prediction studies, PRSs and the gut microbiome comprise promising components of potential future multiomic risk prediction36,37. It has been previously shown that the gut microbiome and host genetics independently contribute to cross-sectional prediction of host metabolic traits, with improved prediction performance by combining genetics and microbiome over modeling based on host genetics and environmental factors38. However, many previous microbiome studies of disease have retrospective case–control designs, which are susceptible to various selection biases (for example, ascertainment, geographical, demographic biases) as well as technical differences such as sample storage39,40. Prospective studies minimize the risk of many of these biases and enable risk prediction of future disease. Furthermore, the extent to which host genetics and microbiome can jointly predict future risk of common diseases, including their additive value to baseline age and other conventional risk factors, remains unclear.
In the present study, we investigate the predictive capacity of PRSs, the gut microbiome and conventional risk factors for multiple incident common diseases using a population-based prospective cohort. We focus on diseases for which there is prior evidence of substantial predictive capacity for PRSs and the human gut microbiome, that is, coronary artery disease (CAD)12,41, T2D26,42, Alzheimer disease (AD)43,44 and prostate cancer45,46. We utilized the population-based, multiomic FINRISK 2002 cohort47 to assess the individual and combined performance of PRSs, gut microbiome scores and conventional risk factors to incident disease. Finally, we generated and validated multiomic predictive models for each disease and have made these available to the research community.
Results
For those in FINRISK 2002 with imputed genotypes and gut metagenomic sequencing, there were 333 incident cases of CAD, 579 of T2D, 273 of AD and 141 of prostate cancer over a median follow-up of 17.8 years through electronic health records (EHRs). Characteristics of the study sample of FINRISK 2002 cohort for each disease are given in Table 1. For CAD, T2D and AD, baseline clinical risk factors were significantly different between incident cases and non-cases with the exception of smoking for T2D, and sex, diastolic BP (DBP) and HDL for AD. We detected significant differences between case and non-case groups in baseline age and smoking for prostate cancer.
PRSs and conventional risk factors
Previously validated PRSs for CAD12 (PGS000018), T2D42 (PGS000036), AD43 (PGS000334) and prostate cancer45 (PGS000662) were obtained from the Polygenic Score Catalog48 (Methods). Cox regression models were used to assess the predictive performance of PRSs and disease-specific conventional risk factors for incident diseases.
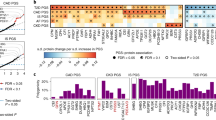
We first assessed prediction performance of PRSs and conventional risk factors (Methods) individually for their respective incident diseases (Fig. 1). In sex-stratified (except for prostate cancer) Cox models of individual risk factors for incident CAD, AD and prostate cancer, baseline age had the highest concordance statistic (C-statistic) (0.719, 95% confidence interval (CI) 0.695–0.743; 0.880, 95% CI 0.864–0.895; and 0.769, 95% CI 0.739–0.798, respectively). For CAD and AD, systolic BP (SBP) was the second strongest individual factor by C-statistics (0.649, 95% CI 0.619–0.679 and 0.656, 95% CI 0.623–0.688, respectively), followed by comparable C-statistics for PRSs (0.626, 95% CI 0.595–0.656 and 0.650, 95% CI 0.616–0.684, respectively). For incident prostate cancer, the PRS was stronger than other individual conventional risk factors except baseline age with a C-statistic of 0.641 (95% CI 0.593–0.690). For incident T2D, the BMI had the strongest C-statistic (0.745, 95% CI 0.726–0.764) and the PRS had a C-statistic of 0.612 (95% CI 0.589–0.636), similar to the other conventional risk factors. The PRS alone achieved a higher C-statistic than family history for all diseases where this was available, including CAD, T2D and prostate cancer.

a–d, C-statistics of Cox models of disease-specific CRFs and PRSs for incident CAD (n = 5,093) (a), T2D (n = 5,297) (b), AD (n = 5,347) (c) and prostate cancer (n = 2,464) (d). CRFs and PRSs are modeled individually and jointly. Cox proportional hazard models for CAD, T2D and AD are stratified by sex. The C-statistics are depicted alongside their 95% CIs as dots and error bars.
In assessing the incremental gain in prediction of each PRS over its disease-specific conventional risk factors (Fig. 1), we found ∆C-indices of 0.023 for CAD (95% CI 0.013–0.034), 0.01 for T2D (95% CI 0.004–0.016), 0.017 for AD (95% CI 0.010–0.024) and 0.027 for prostate cancer (95% CI 0.009–0.047). As expected, all PRSs were significantly associated with their respective incident diseases after adjusting for disease-specific conventional risk factors, and baseline age remained the strongest predictor for CAD, AD and prostate cancer (Extended Data Fig. 1). We observed hazard ratios (HRs) per s.d. for PRS levels of 1.68 for CAD (95% CI 1.50–1.88, P = 2.25 × 10−19), 1.42 for T2D (95% CI 1.30–1.55, P = 6.48 × 10−15), 1.92 for AD (95% CI 1.73–2.15, P = 4.27 × 10−32) and 1.73 for prostate cancer (95% CI 1.47–2.04, P = 5.50 × 10−11). The effects of PRSs and family history were independent for incident CAD, T2D and prostate cancer, implying that the PRS and family history complement each other. As a subanalysis for CAD, we excluded individuals taking antihypertensives and lipid-lowering medications at baseline (Extended Data Fig. 2a,b), with the findings being consistent with the main analysis of all individuals.
For T2D, we performed a subanalysis using nuclear magnetic resonance (NMR)-determined glucose as an additional conventional risk factor (Extended Data Fig. 3a,b). In sex-stratified Cox models of individual risk factors, BMI again had the strongest C-statistic (0.743, 95% CI 0.723–0.764), whereas the PRS and glucose had C-statistics of 0.612 (95% CI 0.588–0.637) and 0.656 (95% CI 0.631–0.682), respectively. Adding the PRS increased the C-statistic over the model of conventional risk factors by 0.007 (95% CI 0.001–0.013). In the model combining PRSs and conventional risk factors, the PRS and glucose were both significantly associated with incident T2D with similar effect sizes (HR = 1.40 per s.d., 95% CI 1.27–1.54, P = 1.85 × 10−12 and HR = 1.38 per s.d., 95% CI 1.28–1.48, P = 5.95 × 10−19).
In a subanalysis of AD in participants aged ≥60 years (Extended Data Fig. 4), the sex-stratified Cox model of the PRS alone with a C-statistic of 0.667 (95% CI 0.629–0.705) was greater than any individual conventional risk factor as well as the model combining all conventional factors. Adding the PRS improved the C-statistic over conventional risk factors by 0.064 (95% CI 0.036–0.096), leading to a model with a C-statistic of 0.722 (95% CI 0.687–0.756). Notably, in the model combining PRSs and all conventional risk factors of AD, the PRS was associated with an incident AD with an HR of 1.87 (95% CI 1.65–2.12, P = 8.95 × 10−23) per s.d., which was greater than that for baseline age (HR = 1.73 per s.d., 95% CI 1.51–1.98, P = 4.50 × 10−15).
Gut microbiome and incident disease
In FINRISK 2002, the gut microbiome composition was determined by shallow shotgun metagenomic sequencing of baseline stool samples (Methods). To investigate the association between incident diseases and the overall variation in gut microbial communities, we performed Cox analyses on α and β diversity at the species level, adjusting for disease-specific conventional risk factors. The α diversity was estimated using the Shannon index, the Chao–Shannon index49, species richness and evenness. The Shannon index and the Chao–Shannon index were significantly negatively associated with incident T2D (HR 0.89 per s.d., 95% CI 0.82–0.96, P = 0.004 and HR 0.90 per s.d., 95% CI 0.82–0.98, P = 0.014, respectively), complementing the previously reported negative association between T2D and gut microbiome richness50; species richness was associated with incident prostate cancer (HR 1.23 per s.d., 95% CI 1.1–1.39, P = 4.20 × 10−4); no significant association was observed for incident CAD and AD (Supplementary Table 1). In the analysis of β diversity between samples using principal component analysis (PCA) of the Aitchison distance, incident T2D was associated with principal component (PC)2 (HR 0.94, 95% CI 0.91–0.96, P = 1.31 × 10−5) and PC5 (HR 1.04, 95% CI 1.00–1.08, P = 0.030). In comparison, using principal coordinate analysis based on the Bray–Curtis dissimilarity, incident T2D was associated with PC1 (HR 1.78, 95% CI 1.08–2.95, P = 0.024) and PC5 (HR 3.26, 95% CI 1.44–7.38, P = 0.005). No significant associations were observed for CAD, AD and prostate cancer.
To investigate the predictive capacity of gut microbial taxa for incident diseases, we focused on 235 species-level taxonomic groups after excluding rare and less prevalent taxa (Methods). In developing prediction models with taxa abundance at species levels, we utilized ridge logistic regression with 10× three-fold stratified cross-validation (Methods). The average cross-validated area under the receiver operating characteristic curve (AUROC) of the models was 0.597 (range 0.588–0.605) for CAD, 0.610 (0.599–0.624) for T2D, 0.564 (0.552–0.582) for AD and 0.613 (0.595–0.626) for prostate cancer (Extended Data Fig. 5). In subanalyses, similar AUROCs of cross-validated models were achieved for CAD (mean 0.587, range 0.552–0.609) and T2D (mean 0.604, range 0.589–0.614), whereas the gut microbiome was not predictive of AD in participants aged ≥60 years at baseline.
In sex-stratified (except for prostate cancer) Cox regression models, the gut microbiome score alone was significantly associated with all incident diseases (Extended Data Fig. 6), with HRs of 1.28 (95% CI 1.17–1.41, P = 2.29 × 10−7), 1.40 (95% CI 1.30–1.51, P = 7.45 × 10−20), 1.34 (95% CI 1.20–1.50, P = 2.09 × 10−7) and 1.50 (95% CI 1.27–1.78, P = 1.66 × 10−6) per s.d. for incident CAD, T2D, AD and prostate cancer, respectively. For CAD and T2D, the gut microbiome scores individually showed similar performance in C-statistics compared with a few conventional risk factors including family history (0.578, 95% CI 0.547–0.61 and 0.612, 95% CI 0.590–0.635, respectively; Fig. 2). For AD, the gut microbiome score achieved a higher C-statistic (0.581, 95% CI 0.546–0.616) than BP, cholesterol levels and smoking. For prostate cancer, the gut microbiome score was second only to baseline age in the C-statistic (0.623, 95% CI 0.581–0.666). After adjusting for disease-specific conventional risk factors (Extended Data Fig. 6), the effect of the gut microbiome score was significant but attenuated for incident T2D (HR = 1.20 per s.d., 95% CI 1.11–1.30, P = 9.13 × 10−6) and prostate cancer (HR 1.23 per s.d., 95% CI 1.03–1.46, P = 0.020); no significant effect of the gut microbiome score was found for CAD and AD. Compared with models of conventional risk factors (Fig. 2), models adding the gut microbiome score yielded a ∆C-statistic of 0.004 (95% CI 0–0.008) for T2D and 0.005 (95% CI −0.003 to 0.013) for prostate cancer. In the subanalysis of T2D using NMR-based glucose as an additional conventional risk factor (Extended Data Fig. 3c), the effect of the gut microbiome score was slightly attenuated (HR 1.16 per s.d., 95% CI 1.07–1.26, P = 5.38 × 10−4) and the ∆C-statistic yielded by adding gut microbiome score to conventional risk factors was 0.003 (95% CI −0.001 to 0.006).

a–d, C-statistics of Cox models of disease-specific CFRs and gut microbial features for incident CAD (n = 5,093) (a), T2D (n = 5,297) (b), AD (n = 5,347) (c) and prostate cancer (n = 2,464) (d). CRFs and gut microbiome scores are modeled individually and jointly. The α diversities and five PCs of CLR abundance are modeled with adjustment for all disease-specific CRFs. Cox proportional hazard models for CAD, T2D and AD are stratified by sex. The C-statistics are depicted alongside their 95% CIs as dots and error bars.
Integrating polygenic, metagenomic and conventional factors
We then investigated the combined predictive performance of PRSs, the gut microbiome and conventional risk factors of their respective diseases using Cox regression models (Table 2). Although age was the strongest individual predictor for incident CAD and prostate cancer, adding the PRS and the gut microbiome score to the age increased the C-statistic by 0.049 (95% CI 0.030–0.066) and 0.032 (95% CI 0.011–0.052), respectively. For T2D, adding the PRS and the gut microbiome score improved the C-statistic over age by 0.076 (95% CI 0.057–0.095). For incident AD, adding the PRS improved the C-statistic over age by 0.019 (95% CI 0.011–0.026), whereas adding the gut microbiome score did not improve the C-statistic. For all four diseases, the model combining disease-specific conventional risk factors, PRSs and gut microbiome scores achieved higher C-statistics than models based on any risk factors separately (Table 2). The combined model achieved ∆C-statistic over conventional risk factors of 0.024 (95% CI 0.013–0.035) for CAD, 0.014 (95% CI 0.007–0.021) for T2D, 0.017 (95% CI 0.009–0.024) for AD and 0.031 (95% CI 0.011–0.05) for prostate cancer.
The subgroup analyses for CAD, T2D and AD showed consistent results in general. In the sex-stratified Cox model for CAD (Extended Data Fig. 2d), adding the PRS and the gut microbiome score increased C-statistics by 0.050 (95% CI 0.030–0.068) over age and 0.025 (95% CI 0.013–0.038) over all conventional risk factors in individuals without baseline use of antihypertensives or lipid-lowering medications. For T2D (Extended Data Fig. 3d), adding the PRS and gut microbiome score improved the C-statistic over age by 0.073 (0.051–0.092) and the combined model increased the C-statistic by 0.010 (95% CI 0.003–0.016) compared with the model of conventional risk factors including NMR-based glucose. In the subgroup analysis for AD in those aged >60 years at baseline, adding the PRS improved the C-statistic over baseline age by 0.077 (95% CI 0.043–0.108), while the gut microbiome score did not show improvement.
In the combined models (Supplementary Tables 2–5), PRSs were found to be significantly associated with CAD (HR per s.d. 1.68, 95% CI 1.50–1.88, P = 4.39 × 10−19), T2D (HR per s.d. 1.41, 95% CI 1.29–1.54, P = 1.38 × 10−14), AD (HR per s.d. 1.93, 95% CI 1.73–2.15, P = 3.85 × 10−32) and prostate cancer (HR per s.d. 1.72, 95% CI 1.46–2.02, P = 1.05 × 10−10). The gut microbiome score was associated with T2D (HR per s.d. 1.19, 95% CI 1.10–1.29, P = 2.11 × 10−5) and prostate cancer (HR per s.d. 1.19, 95% CI 1.01–1.41, P = 0.041).
In subgroup analyses (Supplementary Tables 6–8), similar effects of PRSs were found for CAD (HR per s.d. 1.77, 95% CI 1.56–2.02, P = 3.05 × 10−18), T2D (HR per s.d. 1.40, 95% CI 1.27–1.53, P = 3.43 × 10−12) and AD (HR per s.d. 1.88, 1.65–2.13, P = 8.33 × 10−23); the effect of the gut microbiome score remained significant for T2D (HR per s.d. 1.15, 95% CI 1.06–1.25, P = 1.07 × 10−3) after adjusting for NMR-based glucose and other conventional risk factors.
Discussion
While the interplay between host genetics and the gut microbiome has been increasingly recognized and studied31,51,52, few studies have investigated their combined impact on complex disease risk. The present study presents a joint analysis of genotyping data, gut metagenomics data and clinical metadata for four common complex diseases (CAD, T2D, AD and prostate cancer) in a large prospective population-based cohort. We compared popular published PRSs for each disease, baseline gut metagenomics and conventional risk factors for predicting the onset of each disease over a median of 17.8 years of follow-up. Our analyses reinforce the evidence that baseline age is the dominant individual risk factor for CAD, AD and prostate cancer, and adding the PRS and gut microbiome substantially improved the predictive performance to a similar capacity achieved by the combination of all conventional risk factors. We further demonstrated that PRSs improved prediction performance over the combination of conventional risk factors for all diseases studied, yet there was only mild evidence that the gut microbiome improved prediction performance when modeled jointly with conventional risk factors. The information (for example, features and coefficients) necessary to independently apply our integrated predictive models are provided in Supplementary Tables 2–5.
As expected, in our study, a higher PRS was significantly associated with higher disease incidence for all four diseases, consistent with previous studies. Also expected, we found that PRSs for all four diseases improved predictive ability over conventional risk factors, adding to the body of evidence9,14 that PRSs have potential clinical utility to complement traditional risk factors. Consistent with prior work, we demonstrated that PRSs improved prediction of CAD, T2D and prostate cancer independently of and in addition to family history, a strong risk factor for all diseases studied53,54,55,56,57. Notably, for AD, with the risk of development attributed to genetics being estimated at 70% (ref. 58), the PRS improved the C-statistic over conventional risk factors, including age by 0.017 in all studied participants and 0.064 in participants aged ≥60 years at baseline.
Although the ∆C-statistics for gut microbiome scores over conventional risk factors were small, we observed significant improvement in sex-stratified prediction models over baseline age alone for CAD, T2D and prostate cancer26,59,60,61. In accordance with previous studies, we found a significant inverse signal between baseline α diversity and incident T2D62, which could be partially explained by possible mediation effects of gut microbiota-derived metabolites correlating with lower microbial diversity (for example, imidazole propionate) and insulin resistance63,64. We also found significant associations between β diversity and incident T2D, which might indicate a shift in microbiome composition involved in disease pathogenesis and progression26,65,66.
Our results suggest that the physiological and metabolic processes influenced by risk-associated changes in the gut microbiome vary across diseases. For CAD and T2D, the gut microbiome score exhibited predictive performance comparable to SBP, cholesterol levels and triglycerides. For CAD, AD and prostate cancer, the microbiome score’s predictive effects were largely captured by baseline age; however, this was true to a lesser extent with T2D (Extended Data Fig. 6). The variability in the predictive capacity of the gut microbiome might be partially explained by the reciprocal relationship between host aging and microbial alterations, where age-related and disease-related changes of gut microbiota bidirectionally interact with age-related diseases such as CAD, AD and prostate cancer67.
Our study has limitations. First, the gut microbiome and conventional risk factors were measured only once at the initial assessment. Although the gut microbiome remains largely stable during adulthood, the microbial community is influenced by environment and cohabitation in the long term38,68,69; thus their effects on future disease may change from what we estimated here. In particular, the assessment of predictive capacity for the gut microbiome might be hindered by the overlapping nature of changes in the microbiome and aging-related processes that lead to disease67. Second, owing to unavailability, we did not assess the impact of family history of AD, a risk factor that may also capture important aspects of shared environment influencing gut microbiome composition70,71. Third, the generalizability of the microbiome and integrated risk models to other external cohorts could not be investigated owing to the paucity of large prospective studies with similar data types. The composition of the human gut microbiome differs across geographically and culturally distinct settings, which can be attributed to variations in host genetics, immunity and behavioral features72,73. Last, our study cohort comprised European ancestry (Finnish) participants; thus predictive performance of the PRS and improvement over conventional risk factors may not generalize to other demographics and healthcare systems, particularly as the predictive performance of the PRSs derived in Europeans is known to be attenuated when applied to populations of non-European ancestries74,75,76.
In summary, this work presents one of the first studies on prediction of incident common complex diseases integrating PRSs, gut metagenomics and clinical metadata. Our study highlights potential limitations in the use of the human gut microbiome for improving clinical risk prediction despite its association with incident disease; however, larger studies are warranted to better quantify potential incremental gains. Overall, we show that integrating PRSs and gut metagenomic scores can maximize predictive capacity for common diseases over conventional risk factors alone.
Methods
Study design
The FINRISK surveys have been conducted to investigate risk factors for major chronic noncommunicable diseases every 5 years since 1972 in Finland77. This work was based on the FINRISK 2002 cohort, which contains metagenome data linked to comprehensive metadata at a baseline clinical visit and prospective follow-up and has been studied for the association between gut microbiota and various health outcomes4,26,28,29,31,78. The study included independent and representative population samples of six geographical areas of Finland: (1) North Karelia, (2) North Savo, (3) Turku and Loimaa, (4) Helsinki and Vantaa, (5) Oulu and (6) Lapland; these were randomly drawn from the National Population Information System47. With an overall participant rate of 65%, the FINRISK 2002 cohort comprised a total of 8,783 individuals, including both men and woman, out of 13,498 invitees aged 25–74 years. The participants filled in self-administered questionnaires, undertook health examinations conducted by trained personnel at the study sites and donated biological samples including venous blood and stool. All participants gave written informed consent and the study protocol was approved by the Coordinating Ethics Committee of the Helsinki University Hospital District (ref. no. 558/E3/2001). The FINRISK participation was voluntary and no financial compensation was paid. The surveys were conducted in accordance with the World Medical Association’s Declaration of Helsinki on ethical principles. In the present study, we included individuals whose genotyping data and shotgun metagenomics sequencing of stool samples were both available. We excluded individuals with (1) low reads of metagenomic sequencing (total mapped reads <100,000), (2) baseline pregnancy, (3) BMI ≤40 kg m−2 or <16.5 kg m−2 and (4) antibiotic use up to 1 month before baseline. Altogether, samples from 5,676 participants were eligible for the present study.
Baseline examination and sample collection
Demographic factors, physiological measurements, lifestyle factors, biomarkers and biological samples were collected at baseline in 200247. Questionnaires and invitation to health examinations were mailed to all subjects. Self-administered questionnaires included information such as participant’s background, medical history, diet and self-reported family history of some diseases. Questionnaires were in paper form and saved to electronic format. The health examination and blood sampling were performed by trained nurses at local health centers or other survey sites. Physical measurements such as weight, height and BP were obtained during the health examination. Venous blood samples were collected for the full cohort. The samples were collected after the participants were fasted for ≥4 h and centrifuged at the field survey sites. The fresh samples were transferred daily to the central laboratory of the Finnish Institute for Health and Welfare and analyzed over the next 2 days.
Stool samples were collected from willing participants at home by using an ad hoc kit constructed in-house at the Finnish Institute for Health and Welfare with detailed instructions and a scoop method. The participants were advised to collect the sample preferably in the morning, but any time convenient to the participant was considered acceptable. The samples were mailed overnight between Monday and Thursday to the laboratory of the Finnish Institute for Health and Welfare under winter conditions in Finland and immediately stored at −20 °C on receipt to minimize potential effects of temperature on variation in microbiome composition79. Special care was taken to avoid delayed transit at the post office over the weekend. The sample collection was done under winter conditions with average temperatures well below 0 °C in Finland from January 2002 to March 2002, and no special arrangements were made with regard to the temperature during transportation. Although possible short-term exposure of samples to room temperature after collection may result in slight variations in the detection and relative abundances of rare taxa80, these variations are relatively minor considering the low environmental temperatures and the primary focus of the present study on common taxa. The stool samples were kept unthawed until 2017 when they were transferred to the University of California San Diego for sequencing.
Disease endpoints, exclusion criteria and factors
We studied four incident diseases: CAD, T2D, AD and prostate cancer. The participants were followed up until 31 December 2019 using EHR linkage to the Finnish national registries. Disease cases were identified based on International Classification of Diseases (ICD)81 codes, Anatomical Therapeutic Chemical (ATC) codes, from the Care Register for Health Care (hospital discharges and specialized outpatient care), Finnish Cancer Register and the Drug Reimbursement and Purchase Registers. CAD cases were defined by ICD-10 I20.0|I21|I22, ICD-9 410|4110, ICD-8 410|4110; T2D cases were defined by ICD-10 E1 (refs. 1,2,3,4), ICD-9 250, ICD-8 250, Kela drug reimbursement code 215 and ATC A10B; AD cases were defined by ICD-10 G30|F00, ICD-9 331.0, ICD-8 290.10, Kela reimbursement code 307, reimbursement with ICD code G30|F00|3110 and ATC N06D; prostate cancer cases were identified in the Finnish Cancer Register. Follow-up time was extracted from EHRs and determined by the years to the first incident event, or death, or end of the follow-up study period.
The conventional risk factors for CAD were defined as follows: age, sex, BMI, SBP, total cholesterol, HDL-cholesterol, current smoking status, exercise, any prevalent diabetes and parental history of myocardial infarction12. Smoking status was defined as current use of tobacco products at baseline. Exercise was defined as regular exercise for at least 3 h per week or regular competitive sports training according to responses to self-administered questionnaires. Individuals with missing values of risk factors were excluded. Individuals with prevalent diagnosis of heart diseases were excluded. A total of 5,093 individuals were considered for CAD analyses. In the subanalysis of CAD, participants with baseline use of antihypertensives or lipid-lowering medications were further excluded, resulting in a subset of 4,293 individuals.
For T2D, the risk factors included age, sex, BMI, SBP, total cholesterol, HDL, triglycerides, current smoking status, exercise and parental history of any diabetes26,54. After individuals with incomplete values of risk factors, any prevalent diabetes, baseline use of diabetes medication and HbA1c (if available) ≥6.5% were excluded, a total of 5,297 individuals were involved in T2D analyses. In an additional subanalysis of T2D, baseline glucose determined by the Nightingale Health NMR platform from frozen serum samples was included as an additional risk factor in a subset of 4,911 individuals.
For AD, the risk factors included age, sex, BMI, SBP, DBP, total cholesterol, HDL, average weekly alcohol consumption, current smoking status, exercise, prevalent T2D, prevalent stroke and any prevalent psychiatric disorders including depression, bipolar disorder and schizophrenia82. We excluded individuals with missing values of risk factors and prevalent dementia, which resulted in 5,347 individuals for analyses of AD. The subanalysis of AD in participants aged ≥60 years at baseline included 1,220 individuals.
For prostate cancer analyses, the risk factors included age, BMI, average weekly alcohol consumption, exercise, current smoking status and parental history of any cancer83. Only male participants were studied. After individuals with incomplete risk factors and prevalent diagnosis of prostate cancer have been excluded, a total of 2,464 individuals remained for analyses of prostate cancer.
Characterization of gut microbiome
DNA extraction was performed using the MagAttract PowerSoil DNA kit (QIAGEN) and the Earth Microbiome Project protocols84. The library generation was carried out with a miniaturized version of the Kapa HyperPlus Illumina-compatible library prep kit (Kapa Biosystems)85. The DNA extracts were normalized to 5 ng of total input per sample using an Echo 550 acoustic liquid-handling robot (Labcyte Inc.). Enzymatic fragmentation (1/10 scale), end-repair and adapter-ligation reactions were performed using a Mosquito HV liquid-handling robot (TTP Labtech Inc.). Sequencing adapters were based on the iTru protocol86, where short universal adapter stubs are ligated first followed by addition of sample-specific barcoded sequences in a subsequent PCR step. Amplified and barcoded libraries were quantified by the PicoGreen assay and sequenced on an Illumina HiSeq 4000 instrument to an average depth of ~900,000 reads per sample. The stool shotgun sequencing was successfully performed in 7,231 individuals. Adapters and low-quality sequences were trimmed with Atropos v.1.1.5 (ref. 87) and host reads were removed with Bowtie2 v.2.3.3 (ref. 88) against the human genome assembly GRCh38. The shotgun metagenomic sequences were analyzed with Oecophylla (https://github.com/biocore/oecophylla) based on Snakemake workflow85,89. Stool metagenomes were classified using Kraken2 v.2.1.0 (ref. 90) and a customized index database based on species definitions from 258,406 reference genomes (comprising 254,090 bacterial and 4,316 archaeal genomes) from GTDB release R06-RS202 (27 April 2021)91. Bracken v.2.5.0 (ref. 92) was used to re-estimate abundances after Kraken2 classification. A threshold of 250 reads per taxon was used to define a positive hit, which resulted in 4,026 species identified with a mean prevalence rate of 4.74%. After removing samples with total mapped read counts <100,000 reads per sample, taxonomic profiles from 7,205 individuals were retained for analyses with 698,067 reads per sample median depth, a minimum of 100,082 reads per sample and a maximum of 19,671,923 reads per sample.
Genotype data processing and polygenic score calculation
Genotyping was undertaken using Illumina genome-wide SNP arrays (HumanCoreExome BeadChip, Human610-Quad BeadChip and HumanOmniExpress)56. After samples with ambiguous gender, missingness >5%, excess heterozygosity and non-European ancestries had been removed and variants with missingness >2%, Hardy–Weinberg equilibrium P < 1 × 10−6 and minor allele count <3 were excluded, the samples were prephased with Eagle2 v.2.3. A Finnish-population-specific reference panel consisting of 2,690 high-coverage, whole-genome sequencing and 5,092 whole-exome sequencing samples was used with IMPUTE2 v.2.3.2 to perform genotype imputation. Postimputation quality control was applied using PLINK v.2.0. Variants with INFO score <0.7, minor allele frequency <1% and Hardy–Weinberg equilibrium P < 1 × 10−6 were excluded. Samples with missing rate >10% were excluded. A total of 7,967,866 variants and 7,281 samples remained after quality control.
For all diseases studied, we calculated PRSs in the FINRISK 2002 cohort using external summary statistics in the Polygenic Score Catalog48. We considered previously published scores that were developed mainly based on large European populations and did not include FINRISK 2002 participants in their development. The Polygenic Score Catalog IDs of the PRSs for CAD, T2D, AD and prostate cancer were PGS000018 (ref. 12), PGS000036 (ref. 42), PGS000334 (ref. 43) and PGS000662 (ref. 45), respectively. Each PRS was computed by multiplying the genotype dosage of each risk allele at each variant by its weight and summing across all variants in the respective score with PRSice-2 (ref. 93). The final PRSs consisted of 1,396,966 variants for the CAD PRSs, 129,793 for the T2D PRSs, 21 for the AD PRSs and 181 for the prostate cancer PRSs.
Statistics and reproducibility
Cox proportional hazard models stratified by sex were first fit for time on study for each incident disease on each of their respective conventional risk factors and PRSs separately. Next, a model combining disease-specific PRSs and conventional risk factors was fit for each disease. Prostate cancer was obviously studied only in men; its respective analysis did not include sex stratification. The ability of models to distinguish between cases and non-cases was assessed and compared with Harrell’s C-statistic, a performance metric for evaluating model discrimination based on censored survival data. Proportional hazard assumptions were examined by Schoenfeld residuals. HR, 95% CIs and two-sided Wald’s test P values were reported for risk factors. Statistical significance was determined with a P-value threshold of 0.05.
The gut microbiota diversities were measured with species-level abundance data before filtering taxa by relative abundance and prevalence. Rarefaction was not directly performed to avoid loss of data and samples had total mapped reads >100,000 after filtering. The α diversity of the gut microbiome was measured by Shannon’s diversity, chao1 and evenness using raw counts. As the original Shannon index can exhibit bias owing to unobserved taxa, a nearly unbiased estimator of Shannon entropy proposed by Chao et al. using subsampling taxa and extrapolation was implemented49,94,95. The β diversity was estimated separately in samples by applying PCA on centered log ratio (CLR) transformed abundance data, that is, using the Aitchison distance, after disease-specific exclusion criteria were applied. Cox proportional hazard models were fit for time on study for each disease on gut microbiome α diversity and the first five PCs of CLR abundance, adjusting for conventional risk factors and stratified by sex (except for prostate cancer analyses).
We subsequently focused on common and abundant taxa that were detected with a prevalence >1% and relative abundance >0.1% in at least 10% of samples. After excluding rare and less prevalent taxa, 235 species-level taxonomic groups were obtained and CLR transformed for prediction modeling. For each incident disease studied, we evaluated the predictive capacity of the gut microbiome composition using Ridge logistic regression models of species-level CLR abundance with repeated cross-validation (three-fold, repeated ten times) stratified for disease status where the training and testing data were separate in each iteration. The prevalidated predicted values in the testing sets based on the optimal cross-validated models trained on species-level CLR abundances were used as the gut microbiome scores in assessing the association between the gut microbiome and incident disease. The optimal λ value of Ridge models was determined from a grid search space ranging from 0.0001 to 100. The prediction performance was assessed using AUROC. For comparison, random forests were performed using repeated cross-validation with the same resampling of each iteration. Overall, random forests were outperformed by Ridge regression, with average cross-validated AUROC of 0.551 (range 0.540–0.559) for CAD, 0.570 (0.564–0.579) for T2D, 0.542 (0.531–0.560) for AD and 0.562 (0.540–0.577) for PC. For each disease studied, sex-stratified (except for prostate cancer) Cox regression model was fit for time on study on the gut microbiome score by itself and with adjustment of disease-specific conventional risk factors.
Finally, we investigated whether disease-specific PRSs and microbiome scores made independent contributions to predicting disease risk. For each incident disease, sex-stratified (except for prostate cancer) Cox models were fit on disease-specific PRSs and microbiome scores separately and in combination, adjusting for age at baseline; Cox models were also fit on baseline age alone for comparison. Sex-stratified (except for prostate cancer) Cox models were then fit on disease-specific PRSs, gut microbiome scores and conventional risk factors, and compared with Cox models combining disease-specific conventional risk factors. Covariates and their respective coefficients in Cox regression models for all diseases studied are reported in Supplementary Tables 2–8.
Statistical analysis was performed with R v.4.2.1 and v.3.6.0, R packages data.table v.1.14.2, survival v.3.2.13, compositions v.2.0.4, iNEXT v.3.0.0, otuSummary v.0.1.2, caret v.6.0.90, glmnet v.4.1.3 and v.2.0.18, boot v.1.3.28, pROC v.1.18.0, ggplot2 v.3.3.5, gridExtra v.2.3, grid v.4.1.2 and cowplot v.1.1.1. The present study is observational so randomization or blinding does not apply. The survey was a population-based study of individuals drawn from the Finnish National Population Register stratified by geographical area, sex and 10-year age group47. Exclusion criteria based on quality control standards, baseline characteristics of participants and disease-specific factors are detailed in Methods where relevant. Data distribution was assumed to be normal, but this was not formally tested. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications26,29,31.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The FINRISK data for the present study are available with a written application to the THL Biobank as instructed on the website of the Biobank (https://thl.fi/en/web/thl-biobank/for-researchers). A separate permission is needed from FINDATA (https://www.findata.fi/en/) for use of the EHR data. Metagenomic data are available through the European Genome–Phenome Archive (EGAD00001007035). PRSs are available through the Polygenic Score Catalog (https://www.pgscatalog.org). GTDB R06-RS202 is available through http://gtdb.ecogenomic.org. Genome assembly GRCh38 is available at http://genome.ucsc.edu. The models and statistical source data generated in the analysis are provided as Supplementary tables and source data. All other data supporting the findings of the present study are available from the corresponding author upon reasonable request.
Code availability
The codes for the main analyses are deposited at https://github.com/dpredprj/PRS_GMS_prediction.
References
Joshi, A. et al. Systems biology in cardiovascular disease: a multiomics approach. Nat. Rev. Cardiol. 18, 313–330 (2021).
Ritchie, S. C. et al. Integrative analysis of the plasma proteome and polygenic risk of cardiometabolic diseases. Nat. Metab. 3, 1476–1483 (2021).
Wigger, L. et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab. 3, 1017–1031 (2021).
Liu, Y. et al. Early prediction of incident liver disease using conventional risk factors and gut-microbiome-augmented gradient boosting. Cell Metab. 34, 719–730.e4 (2022).
Walker, K. A. et al. Large-scale plasma proteomic analysis identifies proteins and pathways associated with dementia risk. Nat. Aging 1, 473–489 (2021).
Migliozzi, S. et al. Integrative multi-omics networks identify PKCδ and DNA-PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy. Nat. Cancer 4, 181–202 (2023).
Lambert, S. A., Abraham, G. & Inouye, M. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet. 28, R133–R142 (2019).
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020).
Polygenic Risk Score Task Force of the International Common Disease Alliance. Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps. Nat. Med. 27, 1876–1884 (2021).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590 (2018).
Klarin, D. & Natarajan, P. Clinical utility of polygenic risk scores for coronary artery disease. Nat. Rev. Cardiol. 19, 291–301 (2022).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Sun, L. et al. Polygenic risk scores in cardiovascular risk prediction: a cohort study and modelling analyses. PLoS Med. 18, e1003498 (2021).
Mavaddat, N. et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 104, 21–34 (2019).
Green, H. D. et al. Applying a genetic risk score for prostate cancer to men with lower urinary tract symptoms in primary care to predict prostate cancer diagnosis: a cohort study in the UK Biobank. Br. J. Cancer 127, 1534–1539 (2022).
Sharp, S. A. et al. Development and standardization of an improved type 1 diabetes genetic risk score for use in newborn screening and incident diagnosis. Diabetes Care 42, 200–207 (2019).
Dornbos, P. et al. A combined polygenic score of 21,293 rare and 22 common variants improves diabetes diagnosis based on hemoglobin A1C levels. Nat. Genet. 54, 1609–1614 (2022).
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022).
Li, Z. et al. Polygenic risk scores have high diagnostic capacity in ankylosing spondylitis. Ann. Rheum. Dis. 80, 1168–1174 (2021).
Hao, L. et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat. Med. 28, 1006–1013 (2022).
Jie, Z. et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 8, 845 (2017).
Meijnikman, A. S. et al. Microbiome-derived ethanol in nonalcoholic fatty liver disease. Nat. Med. 28, 2100–2106 (2022).
Wallen, Z. D. et al. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 13, 6958 (2022).
Reitmeier, S. et al. Arrhythmic gut microbiome signatures predict risk of type 2 diabetes. Cell Host Microbe 28, 258–272.e6 (2020).
Ruuskanen, M. O. et al. Gut microbiome composition is predictive of incident type 2 diabetes in a population cohort of 5,572 Finnish adults. Diabetes Care 45, 811–818 (2022).
Bowerman, K. L. et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun. 11, 5886 (2020).
Liu, Y. et al. The gut microbiome is a significant risk factor for future chronic lung disease. J. Allergy Clin. Immunol. 151, 943–952 (2023).
Salosensaari, A. et al. Taxonomic signatures of cause-specific mortality risk in human gut microbiome. Nat. Commun. 12, 2671 (2021).
Hughes, D. A. et al. Genome-wide associations of human gut microbiome variation and implications for causal inference analyses. Nat. Microbiol. 5, 1079–1087 (2020).
Qin, Y. et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 54, 134–142 (2022).
Lopera-Maya, E. A. et al. Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat. Genet. 54, 143–151 (2022).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159, 789–799 (2014).
Goodrich, J. K. et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19, 731–743 (2016).
Valles-Colomer, M. et al. Variation and transmission of the human gut microbiota across multiple familial generations. Nat. Microbiol. 7, 87–96 (2022).
Patel, A. P. et al. A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease. Nat. Med. 29, 1793–1803 (2023).
Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nat. Rev. Genet. 20, 341–355 (2019).
Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215 (2018).
Geneletti, S., Richardson, S. & Best, N. Adjusting for selection bias in retrospective, case–control studies. Biostatistics 10, 17–31 (2008).
Mann, C. J. Observational research methods. Research design II: cohort, cross sectional, and case-control studies. Emerg. Med. J. 20, 54–60 (2003).
Fromentin, S. et al. Microbiome and metabolome features of the cardiometabolic disease spectrum. Nat. Med. 28, 303–314 (2022).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Zhang, Q. et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 11, 4799 (2020).
Ferreiro, A. L. et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci. Transl. Med. 15, eabo2984 (2023).
Conti, D. V. et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 53, 65–75 (2021).
McCulloch, J. A. & Trinchieri, G. Gut bacteria enable prostate cancer growth. Science 374, 154–155 (2021).
Borodulin, K. et al. Cohort profile: the National FINRISK Study. Int. J. Epidemiol. 47, 696–696i (2018).
Lambert, S. A. et al. The polygenic score catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53, 420–425 (2021).
Chao, A., Wang, Y. T. & Jost, L. Entropy and the species accumulation curve: a novel entropy estimator via discovery rates of new species. Methods Ecol. Evol. 4, 1091–1100 (2013).
Forslund, K. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015).
Xu, F. et al. The interplay between host genetics and the gut microbiome reveals common and distinct microbiome features for complex human diseases. Microbiome 8, 145 (2020).
Priya, S. et al. Identification of shared and disease-specific host gene-microbiome associations across human diseases using multi-omic integration. Nat. Microbiol. 7, 780–795 (2022).
Myers, R. H. et al. Parental history is an independent risk factor for coronary artery disease: the Framingham study. Am. Heart J. 120, 963–969 (1990).
Scott, R. A. et al. The link between family history and risk of type 2 diabetes is not explained by anthropometric, lifestyle or genetic risk factors: the EPIC-InterAct study. Diabetologia 56, 60–69 (2013).
Barber, L. et al. Family history of breast or prostate cancer and prostate cancer risk. Clin. Cancer Res. 24, 5910–5917 (2018).
Mars, N. et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 26, 549–557 (2020).
Huynh-Le, M. P. et al. Polygenic hazard score is associated with prostate cancer in multi-ethnic populations. Nat. Commun. 12, 1236 (2021).
Ballard, C. et al. Alzheimer’s disease. Lancet 377, 1019–1031 (2011).
Tang, W. H. et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584 (2013).
Toya, T. et al. Coronary artery disease is associated with an altered gut microbiome composition. PLoS ONE 15, e0227147 (2020).
Matsushita, M. et al. The gut microbiota associated with high-Gleason prostate cancer. Cancer Sci. 112, 3125–3135 (2021).
Maskarinec, G. et al. The gut microbiome and type 2 diabetes status in the multiethnic cohort. PLoS ONE 16, e0250855 (2021).
Menni, C. et al. Serum metabolites reflecting gut microbiome alpha diversity predict type 2 diabetes. Gut Microbes 11, 1632–1642 (2020).
Chen, Z. et al. Association of insulin resistance and type 2 diabetes with gut microbial diversity: a microbiome-wide analysis from population studies. JAMA Netw. Open 4, e2118811 (2021).
Gurung, M. et al. Role of gut microbiota in type 2 diabetes pathophysiology. eBioMedicine 51, 102590 (2020).
Chávez-Carbajal, A. et al. Characterization of the gut microbiota of individuals at different T2D stages reveals a complex relationship with the host. Microorganisms 8, 94 (2020).
Ghosh, T. S., Shanahan, F. & O’Toole, P. W. The gut microbiome as a modulator of healthy ageing. Nat. Rev. Gastroenterol. Hepatol. 19, 565–584 (2022).
Fassarella, M. et al. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut 70, 595–605 (2021).
Valles-Colomer, M. et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature 614, 125–135 (2023).
Donix, M. et al. Influence of Alzheimer disease family history and genetic risk on cognitive performance in healthy middle-aged and older people. Am. J. Geriatr. Psychiatry 20, 565–573 (2012).
Wells, P. M. et al. Associations between gut microbiota and genetic risk for rheumatoid arthritis in the absence of disease: a cross-sectional study. Lancet Rheumatol. 2, e418–e427 (2020).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Gupta, V. K., Paul, S. & Dutta, C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 8, 1162 (2017).
Duncan, L. et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 10, 3328 (2019).
Kamiza, A. B. et al. Transferability of genetic risk scores in African populations. Nat. Med. 28, 1163–1166 (2022).
Araújo, D. S. & Wheeler, H. E. Genetic and environmental variation impact transferability of polygenic risk scores. Cell Rep. Med. 3, 100687 (2022).
Borodulin, K. et al. Daily sedentary time and risk of cardiovascular disease: the national FINRISK 2002 study. J. Phys. Act. Health 12, 904–908 (2015).
Palmu, J. et al. Gut microbiome and atrial fibrillation—results from a large population-based study. eBioMedicine 91, 104583 (2023).
Choo, J. M., Leong, L. E. X. & Rogers, G. B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 5, 16350 (2015).
Flores, R. et al. Collection media and delayed freezing effects on microbial composition of human stool. Microbiome 3, 33 (2015).
International Statistical Classification of Diseases and Related Health Problems, 10th Revision, 5th edn (World Health Organization, 2016).
Silva, M. V. F. et al. Alzheimer’s disease: risk factors and potentially protective measures. J. Biomed. Sci. 26, 33 (2019).
Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 10, 63–89 (2019).
Marotz, L. et al. Earth Microbiome Project (EMP) high throughput (HTP) DNA extraction protocol. Protocols https://doi.org/10.17504/protocols.io.pdmdi46 (2018).
Sanders, J. G. et al. Optimizing sequencing protocols for leaderboard metagenomics by combining long and short reads. Genome Biol. 20, 226 (2019).
Glenn, T. C. et al. Adapterama I: universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). PeerJ 7, e7755 (2019).
Didion, J. P., Martin, M. & Collins, F. S. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5, e3720 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Köster, J. & Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2021).
Lu, J. et al. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, e104 (2017).
Choi, S. W. & O’Reilly, P. F. PRSice-2: polygenic risk score software for biobank-scale data. Gigascience 8, giz082 (2019).
Willis, A. D. & Martin, B. D. Estimating diversity in networked ecological communities. Biostatistics 23, 207–222 (2020).
Hsieh, T. C., Ma, K. H. & Chao, A. iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 7, 1451–1456 (2016).
Acknowledgements
Y.L. was supported by funding from the Cambridge Baker Centre for Systems Genomics. S.C.R. was supported by a British Heart Foundation program grant (no. RG/18/13/33946). M.O.R. was funded by the Research Council of Finland (grant no. 338818). L.L. was supported by the European Union’s Horizon 2020 research and innovation program (grant no. 952914). T.N. was supported by the Finnish Foundation for Cardiovascular Research, the Sigrid Jusélius Foundation, the Southwestern Finland Hospital District and the Research Council of Finland (grant nos. 321351 and 354447). V.S. was supported by the Finnish Foundation for Cardiovascular Research and the Juho Vainio Foundation. A.S.H. was supported by the Research Council of Finland (grant no. 321356). M.I. was supported by the Munz Chair of Cardiovascular Prediction and Prevention and the NIHR Cambridge Biomedical Research Centre (grant nos. BRC-1215-20014 and NIHR203312). M.I. was also supported by the UK Economic and Social Research 878 Council (grant no. ES/T013192/1). The present study was supported by the Victorian Government’s Operational Infrastructure Support program and by core funding from the British Heart Foundation (grant no. RG/18/13/33946) and the NIHR Cambridge Biomedical Research Centre (grant nos. BRC-1215-20014 and NIHR203312). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. This work was supported by Health Data Research UK, which is funded by the UK Medical Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council, Department of Health and Social Care (England), Chief Scientist Office of the Scottish Government Health and Social Care Directorates, Health and Social Care Research and Development Division (Welsh Government), Public Health Agency (Northern Ireland), British Heart Foundation and Wellcome.
Author information
Authors and Affiliations
Contributions
Y.L. and M.I. conceived and designed the study. Y.L., M.O.R., O.K., Q.Z., J.S., P.J., L.L., T.N., V.S., A.S.H., R.K., G.M. and M.I. contributed to investigation of the cohort study and samples. Q.Z., J.S., Y.V.-B., R.K., G.M. and Y.L. processed and analyzed the metagenomics data. A.S.H. and Y.L. processed and analyzed EHR data. Y.L. developed and performed the modeling pipeline and wrote the original draft. S.C.R., S.M.T., K.V., P.J., L.L., T.N., V.S., A.S.H., R.K. G.M. and M.I. provided critical feedback on the study. Y.L., S.C.R. and M.I. prepared the manuscript with input from all authors and all authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
V.S. has had research collaboration with Bayer Ltd (outside the present study). T.N. has received speaking honoraria from Servier Finland and AstraZeneca (not related to the present study). M.I. is a trustee of the Public Health Genomics (PHG) Foundation and a member of the Scientific Advisory Board of Open Targets and has research collaborations with AstraZeneca, Nightingale Health and Pfizer (not related to the present study). The other authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Significant associations between PRSs and incident diseases.
Cox proportional hazards models of disease-specific PRSs and conventional risk factors are fit for (a) CAD (n = 5,093), (b) T2D (n = 5,297), (c) AD (n = 5,347) and (d) prostate cancer (n = 2,464). Cox models for CAD, T2D and AD are stratified by sex. Hazard ratios (HRs) of risk factors are depicted alongside their 95% confidence intervals (CIs) as dots and error bars.
Extended Data Fig. 2 Sub-analysis of incident CAD in individuals who were not on antihypertensives and lipid-lowering medications at baseline (n = 4,293).
In sex-stratified Cox models of PRS and conventional risk factors, (a) C-statistics and (b) hazard ratios (HRs) are depicted alongside their 95% confidence intervals (CIs) as dots and error bars. In sex-stratified Cox models of the gut microbiome score and conventional risk factors, (c) HRs of the gut microbiome score and conventional risk factors are depicted alongside their 95% CIs as dots and error bars. (d) In Cox models for integrative analysis, C-statistics and their 95% CIs of are presented as dots and error bars.
Extended Data Fig. 3 Sub-analysis of incident T2D (n = 4,911) using NMR-determined glucose as an additional risk factor in sex-stratified Cox models.
In sex-stratified Cox models of PRS and conventional risk factors, (a) C-statistics and (b) hazard ratios (HRs) are depicted alongside their 95% confidence intervals (CIs) as dots and error bars. In sex-stratified Cox models of the gut microbiome score and conventional risk factors, (c) HRs of the gut microbiome score and conventional risk factors are depicted alongside their 95% CIs as dots and error bars. (d) In Cox models for integrative analysis, C-statistics and their 95% CIs of are presented as dots and error bars.
Extended Data Fig. 4 Sub-analysis of incident AD in participants aged 60 and above at baseline (n = 1,220) using sex-stratified Cox models of conventional risk factors and PRS.
(a) C-statistics and (b) hazard ratios (HRs) are depicted as dots and their 95% confidence intervals (CIs) are depicted as error bars.
Extended Data Fig. 5 Cross-validated Ridge logistic regression models for incident (a) CAD, (b) T2D, (c) AD and (d) prostate cancer using gut microbiome composition.
The ROC curve of the optimal cross-validated model is in red and curves of other models are in grey.
Extended Data Fig. 6 Cox proportional hazards models of disease-specific gut microbiome scores and conventional risk factors for (a) incident CAD (n = 5,093), (b) T2D (n = 5,297), (c) AD (n = 5,347) and (d) prostate cancer (n = 2,464).
The gut microbiome score is modelled individually and in combination with conventional risk factors. Cox models for CAD, T2D and AD are stratified by sex. Hazard ratios (HRs) of risk factors are depicted alongside their 95% confidence intervals (CIs) as dots and error bars.
Supplementary information
Supplementary Tables
Supplementary Table 1: Cox’s regression of α diversities adjusting for disease-specific conventional risk factors of incident diseases. Cox’s models for incident CAD, T2D and AD are stratified by sex. P values for two-sided Wald’s tests. Supplementary Table 2: Sex-stratified Cox’s models of PRSs, gut microbiome scores and conventional risk factors for incident CAD. P values for two-sided Wald’s tests. Supplementary Table 3: Sex-stratified Cox’s models of PRSs, gut microbiome scores and conventional risk factors for incident T2D. P values for two-sided Wald’s tests. Supplementary Table 4: Sex-stratified Cox models of PRSs, gut microbiome scores and conventional risk factors for incident AD. P values for two-sided Wald’s tests. Supplementary Table 5: Sex-stratified Cox’s models of PRSs, gut microbiome scores and conventional risk factors for incident prostate cancer. P values for two-sided Wald’s tests. Supplementary Table 6: Sex-stratified Cox’s models of PRSs, gut microbiome scores and conventional risk factors for incident CAD in subanalysis of individuals who were not taking antihypertensives or lipid-lowering medication at baseline. P values for two-sided Wald’s tests. Supplementary Table 7: Sex-stratified Cox’s models of PRSs, gut microbiome scores and conventional risk factors for incident T2D in subanalysis including NMR-based glucose as an additional risk factor. P values for two-sided Wald’s tests. Supplementary Table 8: Sex-stratified Cox’s models of PRSs, gut microbiome scores and conventional risk factors for incident AD in subanalysis of individuals aged ≥60 years at baseline. P values for two-sided Wald’s tests.
Source data
Source Data Fig. 1
Statistical source data for Fig. 1.
Source Data Fig. 2
Statistical source data for Fig. 2.
Source Data Extended Data Fig. 1
Statistical source data for Extended Data Fig. 1.
Source Data Extended Data Fig. 2
Statistical source data for Extended Data Fig. 2.
Source Data Extended Data Fig. 3
Statistical source data for Extended Data Fig. 3.
Source Data Extended Data Fig. 4
Statistical source data for Extended Data Fig. 4.
Source Data Extended Data Fig. 5
Statistical source data for Extended Data Fig. 5.
Source Data Extended Data Fig. 6
Statistical source data for Extended Data Fig. 6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Ritchie, S.C., Teo, S.M. et al. Integration of polygenic and gut metagenomic risk prediction for common diseases. Nat Aging 4, 584–594 (2024). https://doi.org/10.1038/s43587-024-00590-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-024-00590-7
