
Abstract
Protein representations from deep language models have yielded state-of-the-art performance across many tasks in computational protein engineering. In recent years, progress has primarily focused on parameter count, with recent models’ capacities surpassing the size of the very datasets they were trained on. Here we propose an alternative direction. We show that large language models trained on codons, instead of amino acid sequences, provide high-quality representations that outperform comparable state-of-the-art models across a variety of tasks. In some tasks, such as species recognition, prediction of protein and transcript abundance or melting point estimation, we show that a language model trained on codons outperforms every other published protein language model, including some that contain over 50 times more parameters. These results indicate that, in addition to commonly studied scale and model complexity, the information content of biological data provides an orthogonal direction to improve the power of machine learning in biology.
Similar content being viewed by others
Main
Pretrained language models have become indispensable tools across many areas of computational protein engineering1. Most labelled protein datasets have limited size, therefore vast deep neural networks are first pretrained on a large, unlabelled corpus of sequence information, such as UniRef2, with a self-supervised reconstruction objective. Self-supervised training endows the latent variables of the model with highly informative features, known as learned representations, which can then be leveraged in downstream tasks where limited training data is available. Learned protein representations are currently central to the state-of-the-art tools for predicting variant fitness3,4,5,6, protein function7,8, subcellular localization9, solubility10, binding sites11, signal peptides12, posttranslational modifications13, intrinsic disorder14 and others15,16, and they have shown promise in the path towards accurate alignment-free protein structure prediction17,18,19,20,21. Improving learned representations is therefore a potential path to deliver consistent, substantial improvements across computational protein engineering.
Pathways towards more informative representations have hitherto followed two main directions. Methods have pursued the model of augmented scale, where increasing model capacity monotonically increases performance22. While initial language models reached tens of millions23 or hundreds of millions24 of parameters, later developments have seen models with over 5 billion weights19,25,26 with parameter counts exceeding the size of the training set. Improvements to model architecture have also consistently delivered performance gains. For example, the use of the T5 architecture in ProtTrans displayed consistent improvements in performance over the basic BERT model8,26. The state-of-the-art fitness prediction method, Tranception, modifies the attention mechanism to explicitly attend to contiguous sequences of amino acids6, increasing robustness and performance on deep mutational scanning benchmarks. Both directions are costly in human and computer time, require notable optimization and appear to provide diminishing (logarithmic) returns.
An alternative pathway to improve learned representations may be to use biological data containing richer signals. While protein language models have so far focused on amino acid sequences, there is additional information contained in the DNA sequence encoding the protein. The language of protein-coding DNA (cDNA) relies on 64 nucleotide triads, known as codons, each of which encodes a specific amino acid or the end of a sequence. Although this 64-codon alphabet is highly degenerate, with most amino acids being encoded by up to six different codons, current research suggests that codons encoding the same amino acid (synonymous) are not used interchangeably. Synonymous codon usage has been correlated with protein structural features27,28, and nearly 60 synonymous mutations have been linked to human disease29. A recent experiment suggested that most synonymous mutations in yeast are strongly deleterious30, although these results have since been contested31,32. Codon usage has also been linked to protein folding, with ample evidence that changes in the codon sequence affect folding dynamics33,34,35,36, the folding pathway37 and even the amount of correctly folded protein38. This evidence indicates that synonymous codon usage contains valuable biological information, which could be exploited by machine learning models to enhance the signal-to-noise ratio in predictive tasks.
In this work, we demonstrate that pretraining a protein language model on codon sequences, rather than amino acid sequences, leads to informative protein representations that capture crucial biochemical characteristics. We examine the predictive power of these representations in a number of sequence-level prediction tasks, observing that these representations are comparable to, or superior to amino acid representations from similarly sized models. In several tasks, we observe that codon-based representations outperform all published state-of-the-art amino acid representations, including those from models with over 50 times more parameters. We conclude that finding more biologically informative representations of the data is a meaningful direction towards progress in deep protein engineering that does not suffer from the computational onerousness of larger scale, and is notably simpler than—but also complementary to—improved model architectures. The development of language models based on coding DNA paves the way for studying other regulatory properties, such as the effect of the codon sequence encoding a protein, which do not lend themselves well to traditional amino acid language models.
Results
We developed a protein language model trained on protein-cDNA and examined its ability to produce high-quality representations of protein sequences. We relied on the fact that the codon space is surjective, but not injective, to the amino acid space, therefore the former contains an amount of information higher or, at worst, equal to the latter (Fig. 1b). To test this hypothesis, we trained a large language model with 86 million parameters on a dataset of 9 million non-redundant and diverse cDNA sequences identified from whole-genome sequencing (Fig. 1c). We refer to this model as CaLM (codon adaptation language model). The training set was constructed from the European Nucleotide Archive39, with substantial preprocessing to limit redundancy and save computational cost. We also established a heldout dataset consisting of representative sequences from seven model organisms across the tree of life. Details of model architecture, training protocol and dataset preprocessing are given in the Methods section.

a, Current research suggests that model performance may be improved by either increasing the number of parameters or improving the architecture of the model. In this work, we propose a third, orthogonal dimension: the use of data with higher information content, in this case the codon, rather than the amino acid sequence. b, The map between the codon alphabet and the amino acid alphabet is surjective, but not injective, hence there is more information in the codon space. c, Processing of the training data. The original database of 114 million cDNA sequences was divided into species and clustered at the protein level. d, Scheme of the training and heldout datasets. As heldout, we selected 4,358 sequences from seven organisms spanning all kingdoms of life, and removed any sequence with 40% sequence identity or more from the training set.
Codon pLMs capture the biology of the genetic code
We first considered whether the learned representations from the codon language model captures the biochemistry underlying the genetic code. A model that has extracted meaningful representations should recognize the similarity of codons in the same wobble pair, a non-trivial task as the model represents individual codons as integers, with no features indicating nucleotide composition. The embedding should also capture the similarity of codons encoding amino acids with similar chemical behaviour, as do amino acids language models24. We tested these hypotheses by examining the embedding layer in CaLM (Fig. 2b). Dimensionality reduction shows that amino acids with similar behaviour cluster in similar regions of space. Clustering captures biochemical features that are not directly obvious from class labels: for example, the codons encoding alanine (‘hydrophobic’) appear close to glycine (‘special’), which reflects the small side chain both amino acids display. We also observed that pairs of codons that encode the same amino acid, or that are in the same wobble pair, are closer in the original 768-dimensional latent space than others (P < 0.05, two-sided permutation test with n = 107).

a, Architecture of CaLM. The sequence of codons is mapped to a continuous space via a trainable embedding, and passed through 12 layers of transformer encoders and a dense layer. The embedding is reversed at the end of the architecture. b, Structure of the learned embedding space. Codons with similar biochemical properties (as shown by the colours) tend to occupy adjacent regions of space. Codons encoding for the same residue (amino acid single letter codes shown over the points) tend to be closer (P = 0.017, two-sided permutation test n = 107), as do codons in the same wobble group (P = 0.020, two-sided permutation test n = 107). c, Structure of the latent space shown on one-third of the sequences in our heldout dataset. The latent representations are distributed by species. d, The embedding of the sequences in b using ESM2 (ref. 19), showing a lack of structure (Supplementary Fig. 5). e, Accuracy of a nearest-cluster-centre classifier at predicting the species of a sequence of the remaining two-thirds of the heldout dataset. The codon language model is significantly better than any other model (largest P value is 1.8 × 10−5, two-sided Welch’s t-test). t-SNE, t-distributed stochastic neighbours embedding.
We then considered sequence representations of different organisms. In Fig. 2c, we display the embeddings of a third of the heldout dataset, which contains sequences with at most 40% sequence identity to any sequence in the training set. The sequences of prokaryotes Escherichia coli and Haloferax volcanii are significantly separated from their eukaryotic counterparts (P smaller than numerical precision, two-sided Welch’s t-test). The sequences of Saccharomyces cerevisiae and Pichia pastoris, which belong to the same order, appear intermixed. The region at the centre of the plot where sequences from multiple organisms converge is enriched in highly conserved sequences such as ribosomal proteins or enzymes involved in the cell cycle. We also controlled for artefacts of dimensionality reduction by varying parameters and testing multiple algorithms (Supplementary Figs. 3 and 4). We compared this clustered structure with representations from amino acid language models (Fig. 2d and Supplementary Fig. 5), observing a less clear clustering. These findings suggest that codon representations capture richer sequence-level information that is not accessible to amino acid sequences alone.
We then tested the ability of the representations to assign cDNA sequences to species, using a simplified k-nearest centres classifier on the 768-dimensional latent space. Class centres were defined using one-third of the heldout set, and tested on the remaining two-thirds; the results are shown in Fig. 2e. The sensitivity to the choice of dataset splits is analysed in Supplementary Fig. 7. We observe that CaLM’s classification accuracy is almost twice as high as the best amino acid classifiers, and significantly superior (P < 10−5, two-sided Welch’s t-test). Since our model is at the cDNA level, we controlled for the differential GC content across different species40, observing that a logistic regression classifier would only achieve 48% accuracy, comparable to the predictions of amino acid representations. Our results recapitulate the well-known biological fact that usage of synonymous codons varies substantially throughout the tree of life41,42. We interpret these findings as evidence that the latent space in CaLM can capture features of differential codon usage that are not evident in the amino acid sequence.
Taken together, our results indicate that the codon language model can access biological features that are inaccessible to amino acid language models.
Codon pLMs match state-of-the-art property predictors
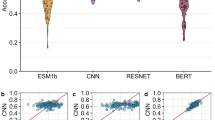
We next examined whether the additional information contained in codon sequences can be used in protein engineering downstream tasks. Several benchmarks of language model representations have been proposed, such as TAPE23, FLIP43 and PROBE8. However, these datasets contain only amino acid sequences, and due to the loss of information, mapping amino acid sequences back to codon sequences is far from trivial. We therefore consider the performance of protein language models in four sequence annotation tasks where it was possible to recover the original codon sequence (Methods): melting point prediction, solubility prediction, subcellular localization prediction and function prediction. These datasets are described in detail in the Methods section. Performance is assessed by fivefold cross-validation (Fig. 3) after subjecting the datasets to a clustering process with tight sequence identity cut-offs (dependent on the dataset, but ranging between 20 and 50%) to ensure removal of homologous sequences. The clustering process ensures that no pair of sequences belonging to different clusters has a sequence identity greater than the threshold, thus ensuring that any machine learning model is not memorizing similar proteins, but achieving some generalization. We also compare the results to two state-of-the-art families of protein language models trained on amino acids: the ESM family of models19,24 and the ProtTrans family of models26, which share a similar architecture to CaLM, have been widely used in protein engineering applications (for example, refs. 5,10) and have achieved top results in protein engineering benchmarks such as FLIP43 or PROBE8.

a, Scheme of the fivefold cross-validation protocol. The data is first divided in five groups such that no pair of sequences belonging to disjoint groups have more than a certain threshold (between 20 and 50%, depending on the dataset) of amino acid identity. In every iteration, a model (a linear or logistic regression) is trained on four groups (green) and tested on the fifth group (red). b–e, The performance of the model with respect to the task and the number of parameters is subsequently shown for melting point prediction (b), solubility prediction (c), subcellular localization classification (d) and function (e) (Gene Ontology term) classification. We report performance using the Pearson correlation coefficient between predicted and true values, or the F1 score averaged over the multiple classes and weighted by class support. Data are presented as mean values with error bars representing the standard deviation calculated from fivefold cross-validation.
We observe that CaLM outperforms every amino acid language model of similar size across all tasks and, in some cases, also amino acid language models with over 50 times more parameters. In melting point prediction, CaLM achieves a Pearson’s R of 0.75 between predicted and experimental values, which is substantially better than any other method in the dataset. In solubility prediction, CaLM outperforms every model of the ESM family with which it shares architecture, and is comparable to the smaller models of the ProtTrans family, which are one order of magnitude larger and trained on two orders of magnitude more data. In subcellular localization and function prediction, the model outperforms all similarly sized architectures and is competitive with many models of greater size and complexity. Performance is commensurate with models hand-crafted for specific tasks, although it is slightly lower than fine-tuned representations. For example, when compared against DeepLoc9, the state-of-the-art tool for subcellular localization prediction, CaLM achieves a weighted F1 score of 0.69 ± 0.02, which is similar to, but still significantly lower than, the DeepLoc score of 0.73 ± 0.01.
We considered the hypothesis that the model may be relying on other information rather than the codon sequence. For example, the model may be learning stability information from species-level signals in the data. Many archaeal proteins are thought to be more stable due to the abundance of ion pairs in their structures44, and since CaLM embeddings can accurately identify the source species of a protein, it might indirectly be using this information in prediction. We controlled for this hypothesis by comparing against an identical version of the linear regression predictor, but including an additional feature that specifies the taxonomic identity of the source species. We observed that CaLM’s predictive power increased from R2 = 0.74 to R2 = 0.78, and that while the absolute difference from the second best method narrowed from six to four percentage points, although it was still significantly better (P = 10−3, two-sided Welch’s t-test). The codon model thus demonstrates superior performance across various unrelated tasks, and against a variety of benchmarks.
We then considered the question of whether the improvement in prediction is due to synonymous codon usage. If patterns of codon usage contain valuable information, then performance should decrease when codon usage is somehow corrupted. We designed an experiment where the results in Fig. 3b were repeated under the same conditions, but randomly mutating a fraction of the codons of both training and test datasets to other codons encoding the same amino acid (synonymous mutations). The results are shown in Supplementary Fig. 8. We observe that Pearson’s R drops from 0.75 to nearly half its value, 0.39, as the sequence of codons is fully randomized, a value that corresponds to the worst performance in the benchmark. To further establish the importance of synonymous mutations, we monitored changes in predictive power when single positions of the protein were mutated to a synonymous variant, observing that the changes were substantially smaller than the inherent variation in the data (Supplementary Fig. 9). These results indicate that the model is extracting useful information from the pattern of synonymous codon usage that is not available from the amino acid sequence.
These findings lead us to conclude that the codon sequence contains valuable information about protein properties that a codon language model is able to extract usefully.
Codon pLMs successfully capture features of omics datasets
We then considered whether our codon language model can be used to predict transcript and protein abundance as measured by transcriptomics and proteomics experiments. We were motivated by the observation that if the codon language model improved performance is due to enhanced biological information, this approach should also be successful on other problems where codon usage is important. The two tasks in this section are related to cDNA composition as codon usage is well-known to present characteristic signatures in housekeeping genes45. To test this hypothesis, we constructed two collections of datasets: of transcript abundance (for the seven organisms, using RNA sequencing (RNA-seq) datasets referenced in Table 1) and protein abundance (for five organisms, as no data was found for H. volcanii or P. pastoris in PAXdb46), and evaluated the ability of CaLM to recover this information using fivefold cross-validation. These datasets are described in detail in the Methods section. We also compared all amino acid-level models, as shown in Fig. 4.

a, Transcript abundance prediction results for the seven organisms represented in our heldout dataset. b, Protein abundance prediction results on five of the seven organisms in our dataset, which were represented in the PAXdb repository46. In both cases, the codon language model shows comparable or better performance to the best language models, and outperforms all language models of similar parameter count. We report performance using the Pearson correlation coefficient between predicted and true values. Data are presented as mean values with error bars representing the standard deviation calculated from fivefold cross-validation.
We observed that the predictions from the codon model yield Pearson’s R2 that are competitive with, or superior to all, amino acid language models for any species. On several species, for example when predicting protein or transcript abundance in the yeasts P. pastoris and S. cerevisiae, the codon language model is substantially superior to any other models. The observation that CaLM can match or outperform amino acid language models, including those that had over 50 times more parameters and were trained on significantly more data, further reinforces the hypothesis that codon language models are able to capture biological features of the sequences that are inaccessible to amino acid language models.
Discussion
In this work, we have shown that protein representations trained on codons, rather than amino acid sequences, exhibit significant advantage across a variety of downstream tasks. We find that our 86 million parameter language model outperforms every other model of similar capacity, and in many cases, even models with over 50 times more parameters. We have provided evidence that this performance is due to the codon language model’s ability to capture patterns of codon usage across DNA sequences, and that this advantage disappears when codon usage information is corrupted.
Training models on cDNA comes at a negligible extra training cost, and appears to increase performance on all sequence-level tasks considered. Since high-throughput protein sequencing is done almost exclusively by translation of DNA sequences, the original coding sequences are publicly available and can be used for training, although they have not been subject to the same standards of processing and annotation as protein sequence databases such as UniRef2. We suggest that using cDNA, instead of simply amino acid sequences, to train protein language models poses a clear pathway towards improving computational protein engineering.
Codon language models may also provide valuable evolutionary signals for alignment-free protein structure prediction, particularly in methods such as ESMfold19 and OmegaFold18 that rely on language models to predict relationships between parts of the protein. Models based on cDNA may recover wider evolutionary relationships, such as synonymous mutations, which are evident at the nucleotide level but not at the amino acid level. Synonymous codon usage is known to relate to structural features27,28, and the connection between codon usage and protein folding33,36 may provide valuable signals to methods that are known to not capture the physics of folding47. We suggest that incorporating codon language models in the pipelines of alignment-free protein structure prediction may well provide a route with negligible cost towards accelerating high-accuracy protein structure prediction.
We propose two main directions towards further improvements in protein representation quality. One is increased scale. The results in this paper have used a simple model with only 86 million parameters, a size that pales in comparison to the standard model size in the latest publications. The dataset used is also relatively small: merely 9 million sequences, in comparison to the 125 million used in the ESM family of models19,24 or the almost half a billion in some ProtTrans models26. There exists a clear pathway towards improving representation quality by training billion-parameter models on datasets comprising hundreds of millions of DNA sequences.
The other potential direction for improvement is the development of multimodal models combining amino acid and coding sequences. Our ablation experiment showed that, in the absence of codon usage information, model performance decays substantially, to the point that it is inferior to every amino acid model in our dataset. However, since the model indirectly has access to the amino acid sequence, it should in principle have access to the same information as amino acid-only models. This divergence may be due to the lack of amino acid-level signals during training, so training models that combine amino acid and codon sequences could improve overall model performance.
The importance of richer data has previously been explored in the domain of applied machine learning. Highly respected papers have shown the importance of higher quality data in vision48, natural language49 and multimodal architectures50. In biology, much attention has been devoted to the impact of dataset biases51, but in comparison little to no attention has been paid to the importance of richer inputs in protein engineering. Our results indicate that, concomitantly with advances in computational power and model architecture, leveraging richer biological data provides a clear direction towards improving the power of machine learning in biology.
The development of large language models trained on cNA will enable the study of properties of the protein that are not directly established by the amino acid sequence. For example, codon usage has been linked to protein folding, with experimental evidence that changes in the codon sequence affect folding dynamics33,34,35,36, the folding pathway37 and even the amount of correctly folded protein38. Careful selection of the sequence of codons is a key objective in protein science, where the specific sequence of cDNA expressed can have a dramatic effect on yield. The codon-based language model presented in this Article represents a first step towards using machine learning methods to study these and other properties of proteins that have hitherto not been addressed by amino acid language models.
Methods
Datasets
Training and test data for unsupervised pretraining
We generated a large corpus of cDNA data to pretrain CaLM using an unsupervised masked language modelling objective. We downloaded the coding sequences of all organisms available in the European Nucleotide Archive with a timestamp of April 2022 (114,214,475 sequences). We considered only high-quality sequences pertaining to assembled genomes (data code ‘CON’). We filtered this dataset to remove all sequences with unknown nucleotides (symbols ‘N’, ‘Y’, ‘R’ and others), with a start codon different from ATG, containing interstitial stop codons or where the number of nucleotides was not a multiple of three. To reduce redundancy while maintaining a representative dataset of codon variation across the tree of life, we grouped the entries by organism, translated the cDNA to protein sequences and clustered the sequences of every organism at 40% amino acid identity using CD-HIT52. We tested that the clustering step did not filter out highly conserved proteins by verifying that nearly all of the members of the Clusters of Orthologous Genes database53 had representative BLAST matches in the dataset (Supplementary Fig. 2). After backmapping clustered sequences to cDNA, the full dataset consisted of 9,858,385 cDNA sequences.
To enable rigorous testing of the model capabilities and generalization power, we built an independent heldout dataset containing sequences of seven model organisms spanning the tree of life: three eukaryotic multicellular organisms (Arabidopsis thaliana, Drosophila melanogaster and Homo sapiens), two eukaryotic unicellular organisms (S. cerevisiae and P. pastoris), a bacteria (E. coli) and an archaea (H. volcanii). We queried GenBank for all cDNA sequences of every model organism according to the highest-quality assembly available, clustered them at 40% amino acid identity and sampled 7.5% of the clustered sequences using random sampling stratified by protein abundance. Since no proteomic data was available for all organisms, we used transcript abundance measured by RNA-seq as a proxy for protein abundance (see Table 1 for data sources). Since we want the heldout dataset to be sufficiently dissimilar from the training set, we used nucleotide BLAST to identify training set sequences with 40% sequence identity or higher to any sequence of the heldout set and removed them. After removing homologous sequences, the training set consisted of 8,771,938 sequences and the heldout of 4,358 sequences.
Evaluation datasets
To test the quality of the representations, we constructed several datasets to test the predictive performance of the learned representations. These datasets overlap with many published benchmarks of learned protein representations. With the exception of the transcriptomics dataset, where the sequence of codons can be inferred from the transcript, all available datasets reported only amino acid sequences. To obtain codon information, we mapped UniProt IDs to European Nucleotide Archive entries using UniProtKB54 and ignored all entries without a match. We also removed all sequences with unknown nucleotides, containing interstitial stop codons or where the number of nucleotides was not a multiple of three.
Melting temperature
We assess the ability of learned representations to predict protein stability using the database of melting temperatures reported in the FLIP set of benchmarks43. This dataset was constructed from a collection of proteome-wide denaturation experiments reported in the Meltome Atlas55. Measured melting temperatures range between 30 and 90 °C, with most of the support in the range between 30 and 55 °C. We used the same splits and homology removal protocol as in FLIP43, where data was clustered at 20% sequence identity.
Subcellular localization
We assess the ability of learned representations to identify the target location of a protein in the cell using the SwissProt localization dataset9, which is also part of the FLIP set of benchmarks43. The SwissProt localization dataset contains ten labels, corresponding to extracellular, cytoplasm, cell membrane, endoplasmic reticulum, Golgi apparatus, lysosome or vacuole, mitochondrion, nucleus, peroxisome and plastid. As expected from the type of the dataset, there is substantial variance in class numerosity, ranging from 0.7% of the proteins being present in the peroxisome, to 3% being present in the Golgi, lysosome and/or vacuole or plastid, to 35% in the cytoplasm or 25% in the extracellular. We used the same clustering as the original authors. Although cluster sizes were slightly different due to UniProt IDs that could not be mapped to cDNA sequences, we noted that fold size variance was small enough to justify conserving the original splits.
Solubility
We assess the ability of learned representations to identify soluble proteins using a custom dataset derived from solubility profiling experiments by Sridharan et al.56. As a proxy for solubility, we used the average protein abundance determined in the SDS-treated fraction of the experiment, that is, in the absence of ATP. The target variable is the fold enrichment with respect to the control; 99% of the proteins have an enrichment of ten or less, with about two-thirds of the protein between 0 and 1 (61%) and another third between 1 and 2.5 (34%). We clustered the sequences at 40% amino acid identity using CD-HIT52.
Gene ontology
We assess the ability of learned representations to predict protein function using a Gene Ontology dataset originally published in the PROBE set of benchmark tasks8. The dataset relies on experimental annotations from UniProtKB and/or SwissProt and UniProtGOA and considers gene ontology groups from all three groups of annotations: that is, molecular function, cellular compartment and biological process. We used the original folds and splits, which were clustered at 50% sequence identity.
Transcriptomics
We assess the ability of learned representations to predict transcript abundance using a custom dataset built from RNA-seq data. We collected RNA-seq datasets for all seven model organisms from the Gene Expression Omnibus, the EMBL-EBI Expression Atlas, the primary literature and the Sequence Read Archive (data sources are reported in Supplementary Table 1, the corresponding assemblies of all organisms are reported in Supplementary Table 2). We estimated transcript abundances of all proteins in the assembly in transcripts per million, and mapped these values to the sequences in the heldout dataset. The target variable is the natural logarithm of the transcript count per million, which ranges between −5 and 13, with most of the proteins (90%) contained between 0 and 10. As this transcriptomic data was used to build the heldout dataset, no further clustering was applied.
Proteomics
We assess the ability of learned representations to predict transcript abundance using a custom dataset built from mass spectrometry protein abundance quantification experiments. We queried the Protein Abundance Database (PAXdb)46 for data on the seven model organisms used in this work. Samples for A. thaliana, D. melanogaster, E. coli, H. sapiens and S. cerevisiae. Dataset coverages were greater than 95% for all five organisms except for A. thaliana with 76% coverage. This data was used to assign protein abundances to all proteins in the heldout dataset for these organisms. The target variable is the estimated number of copies per cell annotated in PAXdb, which ranges between 0 and 105, with most proteins (98.5%) contained between 0 and 103 copies per cell.
Model details
Model architecture
CaLM is a language model inspired by the ESM family of architectures19,24 (see Supplementary Fig. 1 for a detailed architectural diagram). The model consists of three parts: a learnable embedding, a stack of transformer encoder layers and a prediction head. The input sequence is a vector of T tokens, integers that each represent a codon or a special character. For example, the number 11 corresponds to the start codon ‘AUG’, whereas the number 68 represents a special character ‘〈mask〉’ used for masking. The alphabet is composed of the 64 codons, plus five special characters: ‘〈mask〉’ for masking, ‘〈cls〉’ indicating the start of a sequence, ‘〈eos〉’ to indicate the end of a sentence, ‘〈pad〉’ for padding and ‘〈unk〉’ for potentially unknown codons. No previous knowledge is given to the model: codons are represented in an abstract manner and there is, for example, no previous knowledge that codons ‘AUG’ (token number 11) and ‘AUA’ (token number 8) differ only on a single nucleotide.
The vector of tokens, with dimensions of [T] is mapped into a learnable latent space of dimension 768 by the embedding layer, leading to a matrix of size [T, 768]. This matrix is then passed through multiple layers of transformer encoders, following the architecture of Devlin et al.57. The central ingredient in the transformer architecture is the scaled dot-product attention operation, which can be described as follows:
In equation (1), Q, K and V are [B, dk]-dimensional matrices referred to as queries, keys and values, respectively, which are projections of the input vectors using learnable linear transformations. The self-attention process is repeated across multiple parallel heads that are later combined. In our work, transformer layers contain 12 attention heads, with dimension 768, leading to dk = 64. Since the multi-head attention layer is equivariant to permutations of the input tokens, we use rotary positional embeddings58 to enable learning of sequential features. The self-attention block is complemented by residual blocks, a feed-forward neural network and layer normalization steps, resulting in a final output with the dimension [T, 768] (see Supplementary Fig. 1 for more details). Following ref. 24, we use prenormalization to increase stability and no dropout24. The feed-forward neural network at the end of the transformer block has dimension 3,072.
The vector at the end of the stack of 12 transformer layers is referred to as a ‘representation’ and is also of size [T, 768]. The representation vector is the main focus of the paper, although the model is trained alongside a language head that predicts the probability of every token at a given position using this representation vector as input. The language head consists of a feed-forward neural network, followed by layer normalization and a product by the inverse learned embedding matrix. In our work, the language head is a simple feed-forward neural network that maps the 768-dimensional latent space of the representation to the number of tokens. The output logits, when transformed via a softmax, provide an uncalibrated probability distribution over codons. The language model is trained using a masked language modelling (MLM) objective57:
For each sequence x, we sample a percentage of the set of positions \({{\mathcal{M}}}\) where the true token at index i is replaced with another token and we independently minimize the negative log likelihood of the true codon given the masked sequence.
Model training
We trained the model using dynamic masking59. In every training batch we masked 25% of the input tokens at random. Of the masked tokens, 80% were substituted by a special token ‘〈mask〉’ indicating masking, 10% were substituted by another codon at random and the remaining 10% were left untouched. Sequences were trimmed to a maximum size of 1,024 tokens, a number that we found empirically to be sufficiently large to enable efficient learning while preserving computational efficiency. This is consistent with other published models, as 96% of all UniParc entries have fewer than 1,024 amino acids24. Sequences larger than 1,024 codons were subsampled at random at every batch. The size of all sequences in every batch was padded to the maximum sequence in the batch.
We trained the model using the AdamW optimizer with a learning rate of 1 × 10−4 and default parameters otherwise. The learning rate was warmed up from 0 to 10−4 during the first 1,000 gradient steps, and subsequently decayed with a cosine function that reaches zero after 120,000 steps. Gradients were accumulated to an effective batch size of 1,000 examples, or approximately 256,000 tokens. To monitor training, 1% of the training set was reserved at random as validation. The model reported in this work was trained on four NVIDIA Quadro RTX4000 GPUs for 40 days (66,000 gradient steps, 14 full epochs). Training was manually stopped after observing no validation loss improvement for 8,000 steps.
Model evaluation
Embedding visualization
We used the t-distributed stochastic neighbours embedding method to reduce the dimensionality of token and sequence embeddings and enable visualization. We used the implementation of t-distributed stochastic neighbours embedding in sci-kit learn v.0.23.2 (ref. 60) with default parameters. To ensure reproducibility, we performed sensitivity analysis on the perplexity hyperparameter, as well as comparisons with an alternative dimensionality reduction, uniform manifold approximation and projection, which are reported in the Supplementary Information. Plots reported in the main text use the default values of the sci-kit learn implementation, as well as a maximum of 10,000 iterations to ensure convergence.
Source prediction
Protein source prediction was benchmarked with a simple nearest-centroid algorithm. We divided the heldout dataset into two splits: parameter estimation (33%) and test (66%). Using the parameter estimation set, we computed the centroid of all sequences corresponding to a given species. At the test stage, we assigned a sequence to a species according to the centroid with the smallest L2 distance.
Property prediction
We tested the models using fivefold cross-validation. Splits were done using sci-kit learn v.0.23.2 with default parameters and shuffling, except in the subcellular localization task where we used the splits published by DeepLoc. Sequence representations were built using the default parameters of each model and mean-pooled. To ensure that machine learning models trained on embeddings with a vast spread of dimensionalities (320 dimensions for ESM2-6, to 5,120 dimensions in ESM2-48), we applied a dimensionality reduction step to a fixed size of 320 dimensions using principal component analysis61. We used elastic regression for all tasks, except function prediction, where we used a support vector machine to follow the results reported in ref. 8.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We have made available, in our website, the training set (http://opig.stats.ox.ac.uk/data/downloads/training_data.tar.gz), the heldout set (http://opig.stats.ox.ac.uk/data/downloads/heldout.tar.gz) and the weights of the trained model (http://opig.stats.ox.ac.uk/data/downloads/calm_weights.pkl). All datasets used for validation of the models presented in this article are available at https://github.com/oxpig/CaLM.
Code availability
The code required to reproduce the results in this study is available at https://github.com/oxpig/CaLM, and also at the CodeOcean capsule submitted alongside this paper62.
References
Ferruz, N. & Höcker, B. Controllable protein design with language models. Nat. Mach. Intell. 4, 521–532 (2022).
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R. & Wu, C. H. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 (2007).
Reeb, J., Wirth, T. & Rost, B. Variant effect predictions capture some aspects of deep mutational scanning experiments. BMC Bioinformatics 21, 107 (2020).
Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. Adv. Neural Inf. Proc. Syst. 34, 29287–29303 (2021).
Marquet, C. et al. Embeddings from protein language models predict conservation and variant effects. Hum. Genet. 141, 1629–1647 (2021).
Notin, P. et al. Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval. In Proc. International Conference on Machine Learning, 16990–17017 (PMLR, 2022).
Littmann, M., Heinzinger, M., Dallago, C., Olenyi, T. & Rost, B. Embeddings from deep learning transfer GO annotations beyond homology. Sci. Rep. 11, 1160 (2021).
Unsal, S. et al. Learning functional properties of proteins with language models. Nat. Mach. Intell. 4, 227–245 (2022).
Thumuluri, V., Almagro Armenteros, J. J., Johansen, A. R., Nielsen, H. & Winther, O. Deeploc 2.0: multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 50, W228–W234 (2022).
Thumuluri, V. et al. NetSolP: predicting protein solubility in Escherichia coli using language models. Bioinformatics 38, 941–946 (2022).
Littmann, M., Heinzinger, M., Dallago, C., Weissenow, K. & Rost, B. Protein embeddings and deep learning predict binding residues for various ligand classes. Sci. Rep. 11, 23916 (2021).
Teufel, F. et al. Signalp 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Indriani, F., Mahmudah, K. R., Purnama, B. & Satou, K. ProtTrans-glutar: incorporating features from pre-trained transformer-based models for predicting glutarylation sites. Front. Genet. 13, 885929 (2022).
Ilzhoefer, D., Heinzinger, M. & Rost, B. Seth predicts nuances of residue disorder from protein embeddings. Frontiers in Bioinformatics 2: 1019597 (2022)
Høie, M. H. et al. Netsurfp-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res. 50, W510–W515, (2022). https://doi.org/10.1093/nar/gkac439
Bernhofer, M. & Rost, B. TMbed: transmembrane proteins predicted through language model embeddings. BMC Bioinformatics 23, 326 (2022).
Chowdhury, R., Bouatta, N., Biswas, S. et al. Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol 40, 1617–1623 (2022).
Wu, R. et al. High-resolution de novo structure prediction from primary sequence. Preprint at bioRxiv (2022). https://doi.org/10.1101/2022.07.21.500999
Lin, Zeming, et al. "Evolutionary-scale prediction of atomic-level protein structure with a language model." Science 379.6637: 1123-1130. (2023)
Ruffolo, J. A. & Gray, J. J. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Biophysical J. 121, 155a–156a (2022).
Weißenow, K., Heinzinger, M. & Rost, B. Protein language-model embeddings for fast, accurate, and alignment-free protein structure prediction. Structure 30(8), P1169-1177 (2022). https://doi.org/10.1016/j.str.2022.05.001
Kaplan, J. et al. Scaling laws for neural language models. Preprint at arXiv arXiv:2001.08361 (2020). https://arxiv.org/abs/2001.08361
Rao, R. et al. Evaluating protein transfer learning with TAPE. Adv. Neural Inf. Processing Syst. 32, 9689–9701 (2019).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Nijkamp, E., Ruffolo, J., Weinstein, E. N., Naik, N. & Madani, A. Progen2: exploring the boundaries of protein language models. Preprint at arXiv arXiv:2206.13517 (2022). https://arxiv.org/abs/2206.13517
Elnaggar, A. et al. ProtTrans: towards cracking the language of lifes code through self-supervised deep learning and high performance computing. IEEE Transactions on Pattern Analysis and Machine Intelligence, 44(10), 7112-7127 (2021)
Saunders, R. & Deane, C. M. Synonymous codon usage influences the local protein structure observed. Nucleic Acids Res. 38, 6719–6728 (2010).
Rosenberg, A. A., Marx, A. & Bronstein, A. M. Codon-specific Ramachandran plots show amino acid backbone conformation depends on identity of the translated codon. Nat. Commun. 13, 2815 (2022).
Lin, B. C., Kaissarian, N. M. & Kimchi-Sarfaty, C. Implementing computational methods in tandem with synonymous gene recoding for therapeutic development. Trends Pharmacol. Sci. 44(2), P73-84 (2022). https://doi.org/10.1016/j.tips.2022.09.008
Shen, X., Song, S., Li, C. & Zhang, J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 606, 725–731 (2022).
Kruglyak, L. et al. No evidence that synonymous mutations in yeast genes are mostly deleterious. Preprint at bioRxiv (2022). https://doi.org/10.1101/2022.07.14.500130
Dhindsa, Ryan S., et al. "A minimal role for synonymous variation in human disease." The American Journal of Human Genetics 109.12: 2105-2109 (2022).
Nissley, D. A. & O’Brien, E. P. Timing is everything: unifying codon translation rates and nascent proteome behavior. J. Am. Chem. Soc. 136, 17892–17898 (2014).
Sander, I. M., Chaney, J. L. & Clark, P. L. Expanding Anfinsen’s principle: contributions of synonymous codon selection to rational protein design. J. Am. Chem. Soc. 136, 858–861 (2014).
Chaney, J. L. & Clark, P. L. Roles for synonymous codon usage in protein biogenesis. Ann. Rev. Biophys. 44, 143–166 (2015).
Liu, Y., Yang, Q. & Zhao, F. Synonymous but not silent: the codon usage code for gene expression and protein folding. Ann. Rev. Biochem. 90, 375 (2021).
Jiang, Yang, et al. "How synonymous mutations alter enzyme structure and function over long timescales." Nature Chemistry 15.3: 308-318 (2023).
Nissley, D. A. et al. Universal protein misfolding intermediates can bypass the proteostasis network and remain soluble and less functional. Nat. Commun. 13, 3081 (2022).
Cummins, C. et al. The European Nucleotide Archive in 2021. Nucleic Acids Res. 50, D106–D110 (2022).
Birdsell, J. A. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol. Biol. Evol. 19, 1181–1197 (2002).
Nakamura, Y., Gojobori, T. & Ikemura, T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292–292 (2000).
Subramanian, K., Payne, B., Feyertag, F. & Alvarez-Ponce, D. The codon statistics database: a database of codon usage bias. Mol. Biology Evol. 39, msac157 (2022).
Dallago, C. et al. FLIP: Benchmark tasks in fitness landscape inference for proteins. Preprint at bioRxiv (2021). https://doi.org/10.1101/2021.11.09.467890
Nelson, D. L., Lehninger, A. L. & Cox, M. M. Lehninger Principles of Biochemistry (Macmillan, 2008).
Sharp, P. M. & Li, W.-H. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295 (1987).
Wang, M. et al. PAXdb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteom. 11, 492–500 (2012).
Outeiral, C., Nissley, D. A. & Deane, C. M. Current structure predictors are not learning the physics of protein folding. Bioinformatics 38, 1881–1887 (2022).
Sun, C., Shrivastava, A., Singh, S. & Gupta, A. Revisiting unreasonable effectiveness of data in deep learning era. In Proc. IEEE International Conference on Computer Vision, 843–852 (IEEE, 2017).
Hoffmann, J. et al. Training compute-optimal large language models. Preprint arXiv arXiv:2203.15556 (2022). https://arxiv.org/abs/2203.15556
Hendricks, L. A., Mellor, J., Schneider, R., Alayrac, J.-B. & Nematzadeh, A. Decoupling the role of data, attention, and losses in multimodal transformers. Trans. Assoc. Comput. Linguist. 9, 570–585 (2021).
Klarner, L., Reutlinger, M., Schindler, T., Deane, C. & Morris, G. Bias in the benchmark: systematic experimental errors in bioactivity databases confound multi-task and meta-learning algorithms. In Proc. ICML 2022 2nd AI for Science Workshop (2022). https://openreview.net/forum?id=Gc5oq8sr6A3
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Galperin, M. Y., Kristensen, D. M., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Microbial genome analysis: the COG approach. Brief. Bioinformatics 20, 1063–1070 (2019).
Breuza, L. et al. The UniProtkb guide to the human proteome. Database, bav120 (2016). https://doi.org/10.1093/database/bav120
Jarzab, A. et al. Meltome atlas—thermal proteome stability across the tree of life. Nat. Methods 17, 495–503 (2020).
Sridharan, S. et al. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 10, 1155 (2019).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. Preprint arXiv arXiv:1810.04805 (2018). https://arxiv.org/abs/1810.04805
Su, J., Lu, Y., Pan, S., Wen, B. & Liu, Y. Roformer: enhanced transformer with rotary position embedding. Preprint at arXiv arXiv:2104.09864 (2021). https://arxiv.org/abs/2104.09864
Liu, Y. et al. Roberta: a robustly optimized BERT pretraining approach. Preprint at arXiv arXiv:1907.11692 (2019). https://arxiv.org/abs/1907.11692
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Bishop, C. M. Pattern Recognition and Machine Learning (Springer, 2006).
Outeiral, C. Codon adaptation language model (CaLM) (CodeOcean, 2023).
Acknowledgements
We thank O. M. Crook and T. H. Olsen for enlightening discussions. C. O. thanks the UK’s Engineering and Physical Sciences Research Council for financial support through an Engineering and Physical Sciences Research Council Doctoral Prize (grant no. EP/T517811/1) and a postdoctoral fellowship (grant no. EP/W522582/1). This project is supported by the Eric and Wendy Schmidt AI in Science Postdoctoral Fellowship, a Schmidt Futures programme.
Author information
Authors and Affiliations
Contributions
C.O. and C.M.D. had the original idea for the research project. C.O. designed the experiments, collected the data, trained the models and compiled the benchmarks. Both authors contributed to the analysis of the results and the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2 and Figs. 1–9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Outeiral, C., Deane, C.M. Codon language embeddings provide strong signals for use in protein engineering. Nat Mach Intell 6, 170–179 (2024). https://doi.org/10.1038/s42256-024-00791-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-024-00791-0
This article is cited by
-
Genomic language model predicts protein co-regulation and function
Nature Communications (2024)
