
Abstract
Protein structure prediction pipelines based on artificial intelligence, such as AlphaFold2, have achieved near-experimental accuracy. These advanced pipelines mainly rely on multiple sequence alignments (MSAs) as inputs to learn the co-evolution information from the homologous sequences. Nonetheless, searching MSAs from protein databases is time consuming, usually taking tens of minutes. Consequently, we attempt to explore the limits of fast protein structure prediction by using only primary structures of proteins. Our proposed method, HelixFold-Single, combines a large-scale protein language model with the superior geometric learning capability of AlphaFold2. HelixFold-Single first pre-trains a large-scale protein language model with thousands of millions of primary structures utilizing the self-supervised learning paradigm, which will be used as an alternative to MSAs for learning the co-evolution information. Then, by combining the pre-trained protein language model and the essential components of AlphaFold2, we obtain an end-to-end differentiable model to predict the three-dimensional coordinates of atoms from only the primary structure. HelixFold-Single is validated on datasets CASP14 and CAMEO, achieving competitive accuracy with the MSA-based methods on targets with large homologous families. Furthermore, HelixFold-Single consumes much less time than the mainstream pipelines for protein structure prediction, demonstrating its potential in tasks requiring many predictions.
Similar content being viewed by others


Main
Proteins participate in essentially all biological processes and play critical roles for an organism. The structures of proteins are highly correlated to their functions in biological processes. Determining the protein structures to understand their functions can make considerable contributions to life science.
In recent years, protein structure prediction technologies based on artificial intelligence have made sunstantial progress in prediction accuracy, demonstrating great prospects for the drug and vaccine industry. In particular, AlphaFold2 (ref. 1) has pushed the performance to a new frontier in the challenging 14th Critical Assessment of Protein Structure Prediction (CASP14) (ref. 2), approaching the accuracy of experimental determination methods. Mainstream protein structure prediction pipelines rely heavily on co-evolution information extracted from multiple sequence alignments (MSAs). MSAs can be simply regarded as protein chains similar to the target protein chain in sequence. An MSA is related to the co-evolution information of protein sequences, which is crucial to predicting its structure. However, over-reliance on MSAs becomes the bottleneck of various protein-related tasks. Compared with the time (usually a few seconds) required for model inference in the structure prediction pipeline, searching MSAs is time consuming, costing tens of minutes for a protein. The time-consuming search is destructive in tasks demanding high-throughput requests, such as protein design. In the design of therapeutic proteins, such as peptides and antibodies, large-scale virtual screening is typically used to sift through candidate protein datasets to identify potential drugs that can be further validated for a specific target protein. A precise and efficient protein structure prediction method could potentially accelerate the development of new drugs for treating a variety of diseases.
Consequently, designing an accurate and efficient MSA-free protein structure prediction method to is likely to benefit and accelerate the development of protein studies. We argue that a large-scale protein language model (PLM) can serve as an alternative to the MSAs to learn the co-evolution knowledge for MSA-free prediction. An MSA-based method uses the information retrieval technique to explicitly capture co-evolutionary information of a target protein from the protein sequence databases, while a PLM-based method embeds co-evolutionary information into the large-scale model parameters during training and performs an implicit retrieval through model inference, where the PLM can be regarded as a protein knowledge base3. An MSA-based method is less efficient in retrieving information and depends on the retrieval scheme designed manually. On the other hand, a PLM-based method is more efficient in information retrieval, and the quality of retrieval depends primarily on the model’s capacity or parameter size. The past few years have seen tremendous success of large-scale language models4,5,6 in natural language processing, a field that shares many characteristics with protein study. With an increasing number of model parameters, the capacity for learning language knowledge grows substantially. Using self-supervised learning on large-scale unlabelled proteins, PLMs can reveal the long-range interactions along protein sequences and improve downstream protein-related tasks. Advanced works have attempted to adopt PLMs to enhance the performance of multiple downstream tasks, such as estimating the secondary structures and the functions7,8,9,10. In particular, several studies11,12,13 have attempted to apply PLMs to protein structure prediction. Most works first predict the inter-residue two-dimensional geometry using neural networks and then reconstruct the three-dimensional (3D) structure on the basis of energy minimization, which cannot provide end-to-end 3D structure prediction. Moreover, compared with the geometric learning capability of the Evoformer and Structure modules proposed by AlphaFold, the capacities of the geometric models used by these methods, such as recursive models and residual neural networks, are also unsatisfactory in understanding the co-evolution and spatial relations between the residues in a single sequence.
Inspired by the progress of PLMs and AlphaFold2, we propose an end-to-end MSA-free protein structure prediction pipeline, HelixFold-Single. The model used in HelixFold-Single consists of two major components: a large-scale PLM as the foundation and the essential components from AlphaFold2 for folding. The PLM can encode the primary structure into single representation and pair representation to learn the domain knowledge. The Evoformer and Structure modules from AlphaFold2 are then integrated to process the representation, learn the geometric knowledge and then predict the coordinates of atoms. The two components are connected to give an end-to-end differentiable model. HelixFold-Single contains two training stages. In the first stage, the large-scale PLM is trained with thousands of millions of unlabelled single sequences by the task of masked language prediction. In the second stage, we train the whole model with protein structures composed of experimental ground truth and augmentation structures generated by AlphaFold2.
We compare HelixFold-Single with AlphaFold2 and RoseTTAFold on datasets CASP14 and CAMEO (Continuous Automated Model Evaluation). HelixFold-Single achieves accuracy competitive with that of the other methods on proteins with sufficient numbers of homologous sequences. We also analyse the performance of HelixFold-Single on targets with various numbers of homologous sequences: HelixFold-Single is capable of providing accurate structure predictions on most targets, especially targets with large homologous families. An ablation study comparing PLMs of different sizes demonstrates the importance of the size of the PLM for structure prediction. Furthermore, HelixFold-Single shows great superiority in prediction efficiency when compared with the MSA-based methods and could be applied to protein-related tasks demanding a great number of predictions. Specifically, we investigate HelixFold-Single’s precision on various types of representative protein, including peptides, antibodies and nanobodies, with the aim of assessing its potential for application in therapeutic protein design. Our results suggest that HelixFold-Single performs well in predicting flexible regions of these proteins, highlighting its strengths for such applications.
HelixFold-Single
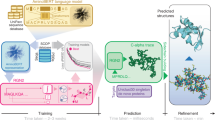
HelixFold-Single aims to take advantage of both the PLM and the main modules used in AlphaFold2 for single-sequence-based protein structure prediction. As exhibited in Fig. 1, HelixFold-Single consists of three components: PLM Base, Adaptor and Geometric Modelling. The large-scale PLM Base is employed to encode the co-evolution information in the parameters, which is used as an alternative to MSAs. Then, in Geometric Modelling, following AlphaFold2, we use modified Evoformer (named EvoformerS) and Structure modules to sufficiently exchange the information between the single representations and pair representations to capture the geometric information and recover the 3D coordinates of the atoms. We adopt an Adaptor layer to extract the co-evolution information from PLM to effectively generate the sequence and pair representations required as inputs to Geometric Modelling. The whole differentiable pipeline is trained by both self-supervised pre-training with bulks of unlabelled single sequences and supervised learning with geometric labels.

It consists of a protein language model as PLM Base, the composite of the EvoformerS (revised from Evoformer) and Structure Module of AlphaFold2 as Geometric Modelling, and Adaptor to connect PLM Base and Geometric Modelling. M, million; K, thousand.
Results
Overall comparison
To compare the overall accuracy of HelixFold-Single with several baseline structure prediction pipelines, including MSA-based and MSA-free methods, we used CASP14 (refs. 1,14,15) with 87 domain targets and CAMEO16 with 371 targets collected from 4 September 2021 to 19 February 2022. AlphaFold2 (ref. 1) and RoseTTAFold17, which rely on MSAs to provide predictions, are currently the most advanced methods for protein structure prediction. We evaluated the prediction performance of AlphaFold2 and RossTTAFold with and without homologous sequences (denoted by AlphaFold2 (input: MSA), RoseTTAFold (input: MSA), AlphaFold2 (input: single) and RoseTTAFold (input: single)). We also trained an MSA-free version of AlphaFold2, denoted by AlphaFold2-Single, by only using the single sequences as input. To evaluate the accuracy of HelixFold-Single and other methods, we utilized a commonly used metric, that is, the template modelling score (TM-score)18.
Figure 2 exhibits the test results of our proposed HelixFold-Single and the compared methods on CASP14 and CAMEO. On the basis of the results, we make the following observations.
-
(1)
In general, HelixFold-Single significantly surpasses all the MSA-free methods on CASP14 and CAMEO and is competitive with the MSA-based methods in certain scenarios. Notably, the accuracy of HelixFold-Single on CAMEO is comparable to that of AlphaFold2 (input: MSA) and outshines another baseline, RoseTTAFold (input: MSA). HelixFold-Single demonstrates the great potential of incorporating PLM into geometric modelling for protein structure prediction.
-
(2)
HelixFold-Single can be on a par with the MSA-based methods on targets with large homologous families, for example, on CASP14 template-based modelling (TBM)-easy domain targets with a median of 7,000 homologous sequences (MSA depth = 7,000) and on CAMEO targets with more than 1,000 homologous sequences (MSA depth > 1,000). These results indicate that the accuracy of HelixFold-Single is correlated to the richness of homologous sequences, revealing that the large-scale PLM adopted by HelixFold-Single is capable of embedding the information, for example, co-evolution knowledge, of MSAs used by the MSA-based methods.
-
(3)
Comparing HelixFold-Single with other MSA-free methods, HelixFold-Single exhibits its great superiority in all the categories of CASP14 and CAMEO. Since AlphaFold2 and RoseTTAFold rely on MSAs as input during the training process, it is challenging for these methods to provide accurate predictions when taking only single sequences as input. Even for AlphaFold2-Single, which uses only single protein sequences as input for training, its precision is unsatisfactory without the assistance of the PLM.

a,b, AlphaFold2 (input: MSA) and RoseTTAFold (input: MSA) are MSA-based methods, while the others use the primary structures as input. Data are divided into quartiles, and a box is drawn between the first and third quartiles, with an additional line drawn along the second quartile to mark the median and a cross to mark the mean. The whiskers extend from the edges of the box to represent the minimum and maximum values within a certain range, excluding outliers. This system is used for all box plots of this paper. a, CASP14 (87 targets classified into free-modelling (FM) and TBM categories on the basis of their relatedness to existing structures.) b, CAMEO (371 targets classified into four categories depending on MSA depth).
Effect of number of homologous sequences
The results on CASP14 and CAMEO indicate that the accuracy of HelixFold-Single is related to the number of homologous sequences. We further compare the performance of HelixFold-Single and other methods on the targets with variant MSA depths. We have collected a fresh test dataset, MSA-Depth-Test, comprising targets that were released between May 2020 and October 2021 from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB). Specifically, we selected targets that exhibit relatively sparse homologous sequences. We blended these targets with the data of CASP14 and CAMEO as a new evaluation set. Figure 3a compares the TM-scores of HelixFold-Single and the baseline methods on the evaluation set, grouped by the number of homologous sequences (MSA depths). Figure 3b shows the distribution of the proteins in different groups in this evaluation set. We can see that as the available homologous sequences grow the average TM-scores of both HelixFold-Single and the MSA-based methods increase, while the scores of the other MSA-free methods decrease. For the proteins with sparse homologous sequences, the TM-scores of all the compared methods are unsatisfactory. For the proteins with larger homologous families, especially those with more than thousands, HelixFold-Single can compete with the MSA-based methods. In general, it appears that HelixFold-Single is more sensitive to the presence of evolutionary information when compared with MSA-based methods such as AlphaFold (input: MSA) or RoseTTAFold (input: MSA). Given that 90% of the targets in PDB have more than 1,024 homologous sequences, we can reasonably extrapolate that HelixFold-Single can achieve satisfying accuracy on the most frequently investigated proteins.

a, Comparison between HelixFold-Single and the baseline methods on 1,251 protein targets with various numbers of homologous sequences (MSA depths). b, Distribution of proteins with different homologous sequences in PDB. c, Relations between MSA depths and TM-scores of HelixFold-Single. d, Relations between MSA depths and perplexity of PLM. e, Relation between perplexity of PLM and TM-scores of HelixFold-Single.
To further investigate the relationship between the capacity of the PLM, the accuracy of protein structure prediction and the size of the homologous family, we utilized the targets in CASP14 and CAMEO datasets to exhibit their relations, as shown in Fig. 3c–e. As we expected, from Fig. 3c, a protein’s structure accuracy (TM-score) is correlated to the size of its homologous family (MSA depth), and the results are consistent with those in Fig. 3b. Moreover, we use a probability metric, perplexity19, to indicate the capacity of the PLM. Perplexity is a metric widely used in natural language processing to quantify the level of uncertainty a language model has in predicting text (which corresponds to the protein sequences in PLM). A lower perplexity score indicates a higher degree of accuracy for the language model. The results in Fig. 3d show that the perplexity of the PLM and the MSA depths are negatively correlated. We reasonably inferred that a PLM would prioritize learning the patterns of high-frequency proteins (which typically have more homologous sequences) rather than long-tail proteins (which usually only have a few homologous sequences) from the large-scale unlabelled protein sequences. These results also explain why the PLM-based HelixFold-Single is more sensitive to MSA depth when predicting protein structures. Moreover, the perplexity of the PLM and the TM-scores of HelixFold-Single are also negatively correlated. These results indicate that if the PLM Base module can predict (model) a protein sequence well, then there is a high probability that the PLM module can learn the co-evolution information of this protein and serves as an alternative to MSAs. Thus, the Geometric Modelling module can leverage the co-evolution embedded in the PLM to provide a more accurate structure for that protein.
Effect of sizes of PLMs
To comprehensively study the ability of the PLMs of different sizes to learn the co-evolution information, we compare a pre-trained PLM of one billion parameters (denoted by PLM-1B) and another pre-trained PLM of 100 million (denoted by PLM-100M). Figure 4a exhibits the perplexity of PLM-1B and PLM-100M on the targets from datasets CASP14 and CAMEO. In general, the smaller the perplexity is, the stronger the capacity of the PLM is. Thus, PLM-1B with more model parameters performs better than PLM-100M with fewer parameters on both datasets CASP14 and CAMEO. In addition, we apply PLM-1B and PLM-100M to the task of protein residue contact prediction to compare their performance on the downstream tasks. We simply fit a logistic regression that takes the attention weights, that is, \([{{{{\boldsymbol{z}}}}}^{(1)},{{{{\boldsymbol{z}}}}}^{(2)},\ldots ,{{{{\boldsymbol{z}}}}}^{({n}_{\mathrm{PLM}})}]\), from the PLMs as input and predict the contact of residues on the targets in datasets CASP14 and CAMEO. Following refs. 7,20, we use the top L/5 long-range contact precision, denoted by P@L/5, where L is the protein length, as the evaluation metric, and the results are shown in Fig. 4b. As we can see, PLM-1B is significantly superior to PLM-100M on the contact prediction task. The results from Fig. 4a and Fig. 4b both support the hypothesis that the larger the size of the PLM, the stronger its capacity. Therefore, it can be reasonably inferred that the performance of the PLM will continue to improve as the size of the PLM increases further.

a, Perplexity of PLM-1B and PLM-100M. b, Contact prediction of PLM-1B and PLM-100M.
Prediction speed comparison
Massive time consumption for searching MSAs is one of the bottlenecks of MSA-based folding, and accelerating the speed of protein structure prediction can considerably broader its applications. The MSA-free HelixFold-Single has a tremendous advantage in inference efficiency by avoiding MSA searching. Figure 5 exhibits the computation time cost of (1) MSA searching, (2) the whole inference pipeline of AlphaFold2 and (3) the inference of HelixFold-Single. All the tests are executed on a single NVIDIA A100(40G) graphics processing unit. In general, HelixFold-Single consumes much less time than AlphaFold2, while the AlphaFold2 pipeline spends most of its time in MSA searching. For proteins less than 100 amino acids in length, HelixFold-Single’s prediction time is only about one-thousandth of that of AlphaFold2. Even for the proteins with more than 800 amino acids, HelixFold-Single still has great efficiency superiority. The good efficiency of HelixFold-Single demonstrates the potential of its application in tasks with a high demand for structural prediction.

We compare the median times of MSA search, AlphaFold2 and HelixFold-Single on proteins with various lengths.
Study on multiple types of representative protein
One of the strengths of HelixFold-Single is its efficiency when compared with MSA-based methods, which makes it well suited for high-throughput protein structure prediction tasks such as protein design. To investigate the performance of HelixFold-Single on therapeutic proteins, three representative types of protein were chosen: peptides, antibodies and nanobodies. Peptides are smaller protein molecules that can be used as drugs to target a variety of biological processes, while antibodies and nanobodies are used in immunotherapy to target specific cells or molecules in the body. An antibody contains two chains, a heavy chain and a light chain, and a nanobody only includes the heavy chain. We evaluate the MSA-free HelixFold-Single and MSA-based AlphaFold2 on multiple datasets—Recent-PDB, Peptide, Antibody and Nanobody—to gain insights into the applicability of these methods to different types of protein and their potential use in protein design. Recent-PDB can be seen as the control group containing recently released proteins from PDB, while the remaining datasets represent experimental groups that are more relevant to therapeutic applications. Antibody-VH and Antibody-VL respectively represent the sets of heavy chains and light chains of collected antibodies.
The results presented in Fig. 6a are intriguing, as they demonstrate that HelixFold-Single can perform as well as, or even outperform, AlphaFold2 in certain scenarios. While HelixFold-Single’s performance slightly lags behind that of AlphaFold2 on the Peptide dataset, the precision gap between the two methods is considerably narrower than that on the Recent-PDB dataset. This indicates that HelixFold-Single is better suited for predicting the structures of short and highly flexible peptides. For the antibody-related datasets, HelixFold-Single performs competitively with AlphaFold2 on datasets Antibody-VL and Nanobody, and surpasses AlphaFold2 on Antibody-VH. We surmise that HelixFold-Single is better equipped to capture the intricate patterns of the complementarity-determining regions (CDRs) from the large-scale protein sequence data, where the CDRs of antibodies are crucial for the specificity of an antibody and are known to be highly variable and difficult to predict. Therefore, we conducted a detailed analysis of HelixFold-Single’s performance on the CDRs, as illustrated in Fig. 6b,c. HelixFold-Single performs comparably to AlphaFold2 in terms of the whole chains (VH, VL and VHH) and all the CDRs, with a slight advantage in predicting the CDR-H3 (widely recognized as the most diverse and critical CDRs) of the antibodies and nanobodies. Given the high variability of short peptides and the CDRs of antibodies, it is reasonable to assume that HelixFold-Single excels in predicting highly variable regions where MSAs may not be effective. To support this hypothesis, we performed additional analyses on the secondary structures of peptides and antibodies. Our results showed that HelixFold-Single is capable of accurately predicting the regions with the more flexible secondary structures of ‘turn’ or ‘coil’. For more information, please refer to Supplementary Section 5.

a–c, Recent-PDB (7,595 targets) is the control group. Peptide (197 targets), Antibody (90 targets) and Nanobody (184 targets) are the sets of representative proteins. Note that a typical antibody has six CDRs, while a nanobody has three CDRs. a, Overall comparison. b, Antibody. c, Nanobody. RMSD, root-mean-square deviation.
Related works
Protein language models
Large-scale language models4 with the self-supervised learning paradigm, such as masked language modelling5 and autoregression21, have achieved extraordinary success in natural language processing tasks. Recent progress has revealed that their capabilities are strongly related to the scale of the model parameters: the larger the scale of the parameters, the better the performance6. The community has not yet seen any sign of growth stopping on moving from billions to hundreds of billions of parameters. These language models are capable of memorizing and generalizing massive common-sense knowledge and professional expertise implicitly included in the large-scale unlabelled data. Inspired by these achievements, PLMs tried to transfer language models and self-supervised learning tasks to protein modelling. A protein can be represented by an amino-acid sequence, similar to the sequences of words or tokens in natural language processing. Previous works7,8,9,10 have shown that, by pre-training with only single sequences without much supervision, PLMs can reveal the protein classification, stability and lower-level structure information (including secondary and tertiary structures and two-dimensional contact maps). However, the accuracy of these models in structure prediction is still far from that of the mainstream folding models supervised by the ground-truth protein structure.
Protein structure prediction
Mainstream pipelines22,23,24,25 rely on extracting the co-evolution information from MSAs to predict the protein structures. Earlier works manually designed the features derived from MSAs, such as inverse covariance matrices. Then, deep neural networks—for example, convolutional networks—are utilized to model the relations between the residues. Advanced studies1,24, directly take the MSAs as input and apply deep neural networks to predict the 3D coordinates of the proteins. In particular, the appearance of AlphaFold2 (ref. 1) has markedly narrowed the accuracy gap between the experimentally determined structures and model-estimated structures, employing the Evoformer module to enhance the interaction between MSA sequences and pairwise geometric information and the Structure module to directly predict the atoms’ coordinates. However, the reliance on MSA inevitably impedes the computation efficiency and accurate prediction of orphan proteins and designed proteins, as well as downstream tasks such as protein design.
Although the structure of a protein is dependent on its primary structure, it is incredibly challenging to train an accurate model that can infer the protein structures with only the primary structures. Only a small number of samples, that is, experimentally determined structures recorded in the PDB database, are available for model training. Several works attempt to incorporate PLMs for MSA-free protein structure prediction. RGN2 (ref. 11) employs a PLM (AminoBERT) with a recurrent geometric network that utilizes Frenet–Serret frames to generate the backbone structure. Moreover, advanced studies12,13 combine pre-trained PLMs, such as ProT5 (ref. 8) and ESM-1b (ref. 26), with residual neural networks to predict two-dimensional structures (for example, a contact map of a protein), yielding superior performance in orphan proteins. Nonetheless, the overall accuracy of those works is still unsatisfactory due to the limited capacity of the model architectures used.
Conclusion and future work
On the one hand, mainstream protein structure prediction methods, such as AlphaFold2 and RoseTTAFold, rely on the MSAs to extract the homologous information. However, searching MSAs is time consuming, limiting the application of those methods to broader protein-related tasks. On the other hand, a large-scale PLM learns the protein correlations from a great number of unlabelled proteins through self-supervised learning tasks. By utilizing large-scale parameters to embed the homologous information, we prove that it can be used as an alternative to MSAs to reduce the time required by the protein structure prediction methods. HelixFold-Single attempts to take advantage of both the PLM and the geometric modelling, predicting the protein structures end to end with only the primary structures. HelixFold-Single can be on a par with the MSA-based methods on targets with large homologous families and is much more efficient than the MSA-based methods, demonstrating its application prospects for protein study.
In the future, as the experimental results indicate that a larger size of PLM can achieve superior performance, we will continue investigating PLMs with a larger size for protein structure prediction. In addition, the accuracy on the targets with only a few homologous sequences is still unsatisfactory. Thus we will try to introduce more diverse training data to alleviate this problem.
Methods
Large-scale PLM Base
Inspired by large-scale pre-trained language models, we follow previous works on pre-training a PLM. The PLM processes the primary protein sequences (that is, the amino-acid sequences) and extracts the knowledge needed for further geometric modelling. A protein of length L can be uniquely represented by a sequence of types of amino acid denoted by x = (x1, x2, …, xL). An embedding layer E(xl) maps the type identifier to dPLM-dimensional embedding vectors:
Notice that \({{{{\mathbf{x}}}}}^{(k)}\in {{\mathbb{R}}}^{L\times {d}_{\mathrm{PLM}}}\) is the representation of the amino-acid sequence.
We then apply the widely used Transformer-style blocks4 to process the embedding vectors, denoted by
Accurately predicting the contacts between the residues, especially the long-rage contacts, is critical for protein structure prediction. Taking into account that the contact between the residues is more dependent on the relative positions rather than the absolute positions (counted from the start of the sequence), we employ DisentangledAttentionTransformer from DeBerTa27 to focus on the modelling of interactions between the residue representations and the relative positions. DisentangledAttentionTransformer adopts the attention mechanism to learn the interactions between the residues as well as the interactions of the interaction–position pairs.
Moreover, we take advantage of multihead self-attention weights in DisentangledAttentionTransformer to construct the initial pair representation. The attention weights of the kth block are denoted by \({{{{\mathbf{z}}}}}^{(k)}\in {{\mathbb{R}}}^{L\times L\times {h}_{\mathrm{PLM}}}\), where hPLM is the number of heads of self-attention.
We add an additional Adaptor to map the output of PLM Base to the input of the Geometric Modelling module.
where nPLM is the number of blocks in PLM Base, and the operator [] refers to concatenation. \({\tilde{{{{\mathbf{x}}}}}}^{(0)}\in {{\mathbb{R}}}^{L\times {d}_{\mathrm{single}}}\) and \({\tilde{{{{\mathbf{z}}}}}}^{({{{\bf{0}}}})}\in {{\mathbb{R}}}^{L\times L\times {d}_{\mathrm{pair}}}\) are the initial single representations and pair representations of the Geometric Modelling module, respectively.
Geometric modelling
We employ the Evoformer and Structure modules proposed in AlphaFold2 (ref. 1) to model the relations between the residues and then estimate the 3D coordinates of the atoms in the proteins. We slightly modify the original Evoformer to match our settings. We name the revised Evoformer EvoformerS (Evoformer with single representations). First, the original Evoformer takes the MSA representation and pair representation, encoded from the searched MSAs, as input. As an alternative, EvoformerS takes the output of Adaptor (including the single representations (\({\tilde{{{{\mathbf{x}}}}}}^{(0)}\)) and pair representations (\({\tilde{{{{\mathbf{z}}}}}}^{(0)}\))). Second, Evoformer adopts various attention mechanisms to exchange the information within the single and pair representations to learn the spatial relationships. Note that, in contrast to the original version of Evoformer proposed by AlphaFold2, we remove the column-wise gated self-attention because HelixFold-Single focuses on MSA-free protein structure prediction and there is no need to exchange the messages within the MSAs. We follow the other geometric components of AlphaFold2, including the Structure module, which takes the single representation and pair representation yielded by EvoformerS and exploits invariant point attention and other geometric transformation operators to predict end to end the 3D coordinates of the atoms. Also, following AlphaFold2, we recycle the whole Geometric Modelling module to refine the predicted structures iteratively.
Model optimization
For the sake of leveraging the domain knowledge from the protein database, we operate two-stage parameter optimization on HelixFold-Single.
In the first stage, the PLM is pre-trained to capture the co-evolution information. The PLM is trained with about 300 million single sequences recorded in a protein database. To encourage PLM to observe the diverse single sequences as soon as possible, we cluster the proteins by similarity of single sequences and sample the proteins to balance the distributions of different clusters in our training data. We apply a self-supervised technique masked language model to optimize the parameters of the PLM, by randomly masking 15% of residues in the single sequences and then reconstructing these masked residues. More concretely, the masked language model attempts to predict P(xl∣x1, …, xl−1, xM, xl+1, …, xL) given the residue in the lth position xl being masked by xM. A crucial proposal of this work is that the PLM can learn the dependence between the masked residue and the other residues, and thus represent the co-evolution information. Previous works7 have already verified that PLMs can reveal secondary structures of the proteins, but the relation between PLM and co-evolution has been little discussed. Co-evolution is the phenomenon that two residues in contact tend to evolve at the same time to preserve the structure and thus the function of the protein. In PLM, if a residue at another position s has a profound impact (if the residue at position s is changed, the masked residue will also change) on the masked residue, then these two residues are likely to evolve at the same time.
In the second stage, since merely relying on PLM to predict the structure is inadequate to capture the geometric information, PLM Base and Geometric Modelling modules in HelixFold-Single are jointly optimized. We utilize 100,000 experimentally determined protein structures. We also use an additional one million estimated protein structures for training in this stage (distilled from AlphaFold2). Following AlphaFold2, we train the network end to end with the main losses, including the frame aligned point error loss and other auxiliary losses. By combining the computationally efficient PLM Base module (compared with MSA search) and the Geometric Modelling module, HelixFold-Single is capable of providing efficient and precise protein structure prediction.
Datasets
We used UniRef30 (2021-03) (ref. 28), which clusters UniRef100 seed sequences from the UniProt Knowledgebase and selected UniProt Archive records29,30 at a 30% pairwise sequence identity level, to pre-train the PLM. Then, three datasets are used to train the whole network, including the proteins in PDB (refs. 31,32) released before 14 May 2020 and two datasets constructed from Uniclust30 (v.2018-08) and AlphaFold Protein Structure Database (v.2022-01) (ref. 33), for knowledge distillation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
To pre-train the PLM, UniRef30 (2021-03) is publicly available at https://wwwuser.gwdg.de/~compbiol/uniclust/2021_03/; to train the whole network, PDB can be downloaded at https://www.rcsb.org/docs/programmatic-access/file-download-services and AlphaFold Protein Structure Database as the distillation dataset can be downloaded at https://ftp.ebi.ac.uk/pub/databases/alphafold/v2/. The CAMEO dataset can be downloaded at https://www.cameo3d.org/modeling/ with dates between 4 September 2021 and 19 February 2022. The CASP14 and CASP15 dataset can be partially downloaded at https://predictioncenter.org/download_area/. The MSA-Depth-Test, Recent-PDB and Peptide sets are filtered from PDB with conditions detailed in Supplementary Information. The Antibody and Nanobody sets can be downloaded at https://opig.stats.ox.ac.uk/webapps/sabdab-sabpred/sabdab/.
Code availability
The source code, trained weights and inference code of HelixFold-Single are freely available at GitHub (https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single) to ensure the reproduction of our experimental results. The version used for this publication is available at ref. 34. A web service of HelixFold-Single is also available at https://paddlehelix.baidu.com/app/drug/protein-single/forecast to provide efficient protein structure predictions.
Data analysis used Python v.3.7, NumPy v.1.16.4 and MMseqs2 release 13-45111. TMscore.cpp v20220227 (https://zhanggroup.org/TM-score/) was used for computing TM-scores.
References
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Moult, J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 15, 285–289 (2005).
Petroni, F. et al. Language models as knowledge bases? In Proc. 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP) https://doi.org/10.18653/v1/D19-1250 (ACL, 2019).
Vaswani, A. et al. Attention is all you need. In NIPS'17: Proc. 31st International Conference on Neural Information Processing Systems Vol. 30 (eds von Luxburg, U. et al.) 6000–6010 (Curran, 2017).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers) (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2019).
Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 33, 1877–1901 (2020).
Rao, R. et al. Evaluating protein transfer learning with TAPE. In NIPS'19: Proc. 33rd International Conference on Neural Information Processing Systems Vol. 32 (eds Wallach, H. M. et al.) 9689–9701 (2019).
Elnaggar, A. et al. ProtTrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. Preprint at arXiv https://doi.org/10.48550/arXiv.2007.06225 (2021).
Rao, R., Meier, J., Sercu, T., Ovchinnikov, S. & Rives, A. Transformer protein language models are unsupervised structure learners. In 9th International Conference on Learning Representations (ICLR, 2021).
Xiao, Y., Qiu, J., Li, Z., Hsieh, C.-Y. & Tang, J. Modeling protein using large-scale pretrain language model. Preprint at arXiv https://doi.org/10.48550/arXiv.2108.07435 (2021).
Chowdhury, R. et al. Single-sequence protein structure prediction using language models from deep learning. Preprint at bioRxiv https://doi.org/10.1101/2021.08.02.454840 (2021).
Weißenow, K., Heinzinger, M. & Rost, B. Protein language-model embeddings for fast, accurate, and alignment-free protein structure prediction. Structure 30, 1169–1177.E4 (2022).
Wang, W., Peng, Z. & Yang, J. Single-sequence protein structure prediction using supervised transformer protein language models. Nat. Comput. Sci. 2, 804–814 (2022).
Kinch, L. N., Schaeffer, R. D., Kryshtafovych, A. & Grishin, N. V. Target classification in the 14th round of the critical assessment of protein structure prediction (CASP14). Proteins 89, 1618–1632 (2021).
Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K. & Moult, J. Critical assessment of methods of protein structure prediction (CASP)–Round XIV. Proteins 89, 1607–1617 (2021).
Robin, X. et al. Continuous Automated Model EvaluatiOn (CAMEO)—perspectives on the future of fully automated evaluation of structure prediction methods. Proteins 89, 1977–1986 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 (2004).
Brown, P. F., Della Pietra, S. A., Della Pietra, V. J., Lai, J. C. & Mercer, R. L. An estimate of an upper bound for the entropy of English. Comput. Linguist. 18, 31–40 (1992).
Rao, R. M. et al. MSA Transformer. Proc. Mach. Learning Res. 139, 8844–8856 (2021).
Radford, A., Narasimhan, K., Salimans, T. & Sutskever, I. Improving language understanding by generative pre-training OpenAI (2018); https://openai.com/research/language-unsupervised
Yang, J. et al. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl Acad. Sci. USA 117, 1496–1503 (2020).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Du, Z. et al. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 16, 5634–5651 (2021).
Peng, J. & Xu, J. RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins 79, 161–171 (2011).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
He, P., Liu, X., Gao, J. & Chen, W. DeBERTa: decoding-enhanced BERT with disentangled attention. In 9th International Conference on Learning Representations (ICLR, 2021).
Mirdita, M. et al. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176 (2017).
Suzek, B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2014).
The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Burley, S. K. et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49, D437–D451 (2020).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2021).
xiaoyao4573 et al. Paddlepaddle/paddlehelix: v1.2.2. Zenodo https://doi.org/10.5281/zenodo.8202943 (2023).
Acknowledgement
This work is supported by the National Engineering Research Center of Deep Learning Technology and Applications.
Author information
Authors and Affiliations
Contributions
X.F., F.W., J.H., X.Z., H.W. and L.S. led the research. L.L., X.F. and F.W. contributed technical ideas. L.L. and D.L. developed the proposed method. Y.X., K.Z. and H.L. developed analytics. X.F., F.W., L.L. and Y.X. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Alexander Pritzel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Jacob Huth, in collaboration with the Nature Machine Intelligence team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Sections A–G, figures, tables and references.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, X., Wang, F., Liu, L. et al. A method for multiple-sequence-alignment-free protein structure prediction using a protein language model. Nat Mach Intell 5, 1087–1096 (2023). https://doi.org/10.1038/s42256-023-00721-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-023-00721-6
This article is cited by
-
Antimicrobial resistance crisis: could artificial intelligence be the solution?
Military Medical Research (2024)
-
Broadening environmental research in the era of accurate protein structure determination and predictions
Frontiers of Environmental Science & Engineering (2024)
-
Protein–protein interaction site prediction by model ensembling with hybrid feature and self-attention
BMC Bioinformatics (2023)
