
Abstract
The development of novel strategies to rapidly construct complex chiral molecules from readily available feedstocks is a long-term pursuit in the chemistry community. Radical-mediated alkene difunctionalizations represent an excellent platform towards this goal. However, asymmetric versions remain highly challenging, and more importantly, examples featuring simple hydrocarbons as reaction partners are elusive. Here we report an asymmetric three-component alkene dicarbofunctionalization capitalizing on the direct activation of C(sp3)–H bonds through the combination of photocatalysed hydrogen atom transfer and nickel catalysis. This protocol provides an efficient platform for installing two vicinal carbon–carbon bonds across alkenes in an atom-economic fashion, providing a wide array of high-value chiral α-aryl/alkenyl carbonyls and phosphonates, as well as 1,1-diarylalkanes from ubiquitous alkane, ether and alcohol feedstocks. This method exhibits operational simplicity, broad substrate scope and excellent regioselectivity, chemoselectivity and enantioselectivity. The compatibility with bioactive motifs and expedient synthesis of pharmaceutically relevant molecules highlight the synthetic potential of this protocol.

Similar content being viewed by others
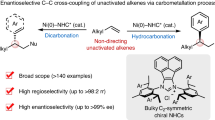
Main
The efficient assembly of high-added-value chiral molecules from readily available, inexpensive hydrocarbons with minimal waste generation represents a long-standing challenge for synthetic chemists1,2. Two major challenges have hampered the broad implementation of these processes: on the one hand, C(sp3)–H bonds present a much lower reactivity compared to most functional groups. Furthermore, the presence of multiple non-equivalent C(sp3)–H bonds requires exquisite control over the chemoselectivity, regioselectivity and stereoselectivity to attain the desired outcome3,4. Recently, hydrogen atom transfer (HAT) photocatalysis has emerged as a mild strategy for direct C(sp3)–H functionalizations5. Upon irradiation with light, the excited state of a photocatalyst can abstract hydrogen atoms from strong and inert C(sp3)–H bonds to produce nucleophilic carbon-centred radicals. Easy-to-synthesize tetra-n-butylammonium decatungstate (TBADT) and diaryl ketones have attracted particular interest as HAT photocatalysts owing to their ability to cleave electronically and sterically accessible C–H bonds with high regioselectivity6,7,8,9. As a result, synthetically useful C(sp3)–H functionalizations promoting the formation of new C–C (refs. 10,11,12,13,14,15,16), C–O (refs. 17,18), C–N (refs. 19,20), C–F (refs. 21,22) and C–S (ref. 23) bonds have been realized capitalizing on this strategy. More recently, the Kong24 and Molander25 groups have independently developed elegant methods for the racemic three-component dicarbofunctionalization of alkenes via photocatalysed HAT (photo-HAT)/nickel dual catalysis.
Catalytic three-component radical alkene difunctionalizations represent a straightforward and efficient approach for the rapid construction of molecular complexity, as two new functional groups can be simultaneously installed across the π system in a single operation26,27,28,29,30,31,32,33,34,35,36. However, controlling the enantioselectivity of the generated stereogenic centres remains a formidable challenge owing to the high reactivity and instability of the open shell radical intermediates participating in these transformations. Chiral copper37,38,39,40,41,42,43 and nickel 44,45,46,47,48,49,50,51,52 complexes have opened a robust platform for asymmetric radical conjunctive cross-coupling of olefins. For example, impressive progress has been made in nickel-catalysed asymmetric radical relayed reductive coupling46,47,48,49, in which C(sp3)-hybridized electrophiles such as alkyl halides and perfluoroalkyl halides have been employed as radical precursors. In addition, processes merging photoredox catalysis and nickel catalysis have enabled the asymmetric redox-neutral dicarbofunctionalization of alkenes with alkyltrifluoroborates as competent synthons for these protocols50,51. All of the aforementioned transformations strongly rely on the use of pre-activated radical precursors, use that not only leads to the generation of additional waste but also results in poor step economy and atom economy. To improve the synthetic efficiency and expand the pool of radical precursors, the direct use of abundant hydrocarbons as coupling partners in the asymmetric dicarbofunctionalization of alkenes represents a more ideal but also highly challenging approach.
Although synergistic HAT processes and transition metal catalysis have been used in the enantioselective arylation, alkylation, alkenylation, acylation and cyanation of C(sp3)–H bonds (Fig. 1a)53,54,55,56,57,58,59,60,61,62,63,64, the direct activation of C(sp3)–H bonds towards the asymmetric difunctionalization of alkenes is certainly underdeveloped (Fig. 1b). Two main challenges need to be addressed: first, the identification of a chiral ligand that can overcome competitive two-component side reactions while efficiently imparting absolute stereocontrol in the formation of the new stereogenic centres in the three-component process; and second, the implementation of reaction conditions wherein no competitive activation of distinct, even more activated C–H bonds present in the solvent and/or the participating reaction partners compromise the efficiency and selectivity of the overall process.

a, Asymmetric two-component C(sp3)–H functionalization through a synergistic HAT process and transition metal catalysis. b, Previous strategies and our design for asymmetric three-component radical dicarbofunctionalization of alkenes. BDE, bond dissociation energy; FG, functional group; PC, photocatalyst; Red, reductant.
Here we show that the combination of a decatungstate or a diaryl ketone HAT photocatalyst with a chiral biimidazoline (BiIm) nickel catalyst enables the asymmetric dicarbofunctionalization of alkenes so that valuable α-aryl/alkenyl carbonyls and phosphonates as well as 1,1-diarylalkanes can be obtained in enantioenriched form through the regioselective activation of abundant hydrocarbons and their derivatives.
Results
Optimization of the reaction conditions
Our investigation to identify suitable reaction conditions commenced with cyclohexane, tert-butyl acrylate and 4-bromobenzonitrile as model substrates under near-ultraviolet light irradiation (Kessil 40 W, 390 nm light-emitting diodes (LEDs); Fig. 2). After extensive evaluation of the reaction parameters, we found that the combination of TBADT (2 mol%), NiBr2·DME (DME, dimethoxyethane; 10 mol%), chiral BiIm L1 (15 mol%) and potassium phosphate (K3PO4, 2 equiv.) provided tert-butyl (R)-2-(4-cyanophenyl)-3-cyclohexylpropanoate 1 in 80% isolated yield and 96:4 enantiomeric ratio (e.r.) in an acetone/trifluorotoluene (PhCF3) binary solvent system at 5 °C (Fig. 2, entry 1). The examination of different ligands revealed that the electronic and steric nature of the chiral BiIm template has a noticeable effect on the reaction efficiency (L1–L5). In general, BiIm ligands bearing electron-withdrawing groups on the aromatic ring result in higher yield but lower enantioselectivity (L4). Electron-rich and sterically demanding substituents on the nitrogen atom are beneficial for imparting stereocontrol, albeit with lower yield (L5). By comparison, 3-(tert-butyl)phenyl-substituted biimidazole L1 exhibited the best compromise between reactivity and enantioselectivity. Reducing the amount of cyclohexane (Fig. 2, entry 2) or ligand (Fig. 2, entry 3), as well as decreasing the loading of nickel/L1 (Fig. 2, entry 4) had a negative effect on the reaction efficiency. The dual acetone/PhCF3 solvent system was essential for the success of this transformation, as a substantial decrease in yield and enantioselectivity was observed when acetone, acetonitrile or CH3CN/PhCF3 was used as solvent (Fig. 2, entries 5–7). Furthermore, replacing the optimal solvent with PhCF3 or acetone/EtOAc resulted in complete failure of the reaction (Fig. 2, entries 8 and 9). Different nickel catalysts such as NiBr2·diglyme and NiBr2·3H2O delivered the product with slightly decreased yield and e.r. (Fig. 2, entries 10 and 11), while NiCl2·DME and NiCl2(Py)4 led to a dramatically reduced yield and enantioselectivity (Fig. 2, entries 12 and 13). The reaction still proceeded smoothly and provided a comparable e.r. upon dilution (Fig. 2, entries 14 and 15). Control experiments confirmed that photocatalyst, nickel, ligand and light were all necessary for a successful outcome (Fig. 2, entry 16). The absolute configuration of product 1 was unambiguously confirmed by X-ray diffraction analysis. In parallel, optimal conditions for isopropanol as radical precursor were sought. Gratifyingly, the desired three-component coupling product 2 could be obtained in 85% yield and 96:4 e.r. in the presence of diaryl ketone PC I, NiBr2·DME, L5 and Na2CO3 under 40 W, 390 nm LED irradiation at 5 °C. The influence of ligand, photocatalyst and solvent is shown in Fig. 2, entries 18–20.

The optimization of reaction conditions. aStandard reaction conditions I are as follows: cyclohexane (2 mmol, 10 equiv.), 4-bromobenzonitrile (0.2 mmol, 1 equiv.), tert-butyl acrylate (0.6 mmol, 3 equiv.), TBADT (0.004 mmol, 2 mol%), NiBr2·DME (0.02 mmol, 10 mol%), L1 (0.03 mmol, 15 mol%), K3PO4 (0.4 mmol, 2 equiv.), acetone/PhCF3 (0.5 ml:0.5 ml), 40 W 390 nm Kessil lamp, N2, 5 °C, 18 h. bStandard reaction conditions II are as follows: isopropanol (2 mmol, 10 equiv.), 4-bromobenzonitrile (0.2 mmol, 1 equiv.), tert-butyl acrylate (0.6 mmol, 3 equiv.), PC I (0.04 mmol, 20 mol%), NiBr2·DME (0.02 mmol, 10 mol%), L5 (0.03 mmol, 15 mol%), Na2CO3 (0.4 mmol, 2 equiv.), acetone/PhCF3 (1.0 ml:1.0 ml), 40 W 390 nm Kessil lamp, N2, 5 °C, 18 h. Isolated yields. The e.r. values were determined by chiral HPLC. cDetected by gas chromatography/mass spectrometry. ND, not detected.
Substrate scope
With the optimized conditions in hand, the scope of this transformation was investigated next. We initially focused on examining a diverse array of aryl/alkenyl bromides (Fig. 3). Aryl bromides bearing cyanide (1), sulfone (3), lactone (4), ketone (5), ester (6), trifluomethyl (7), fluorine (8), chlorine (9), phenyl (11), trifluoromethoxy (12), acetoxy (13), benzoyloxy (14) and phenoxy (15) groups were all successfully converted to the corresponding dicarbofunctionalization products in good yields and high e.r. In general, electron-deficient aryl bromides exhibited improved efficacy compared to electron-neutral (10) and electron-rich ones. Aryl halides bearing strong electron-donating groups, such as 4-bromoanisole and 4-bromothioanisole, were not amenable to the standard reaction conditions. By increasing the light intensity in the reaction, the formation of the corresponding dicarbofunctionalization adducts could be successfully accomplished in these cases (16 and 17). The reactivities and enantioselectivities were not largely impacted by increased steric hindrance, because ortho- and meta-fluoro aryl bromides, as well as 2,4-, 3,4- and 3,5-disubstituted aryl bromides were still compatible with the established protocol (18–23). Polycyclic aryl bromides such as 2-bromonaphthalene, 3-bromo-9H-fluorene and 3-bromophenanthrene were also competent coupling partners (24–26). Moreover, derivatives of flurbiprofen (27) and naproxen (28), two well-known non-steroidal anti-inflammatory drugs (NSAIDs), could be readily synthesized applying our method. Heteroaryl bromides containing thiophene (29), benzothiophene (30), benzofuran (31), dibenzothiophene (32), dibenzofuran (33), benzothiazole (34), pyridines (35), pyrimidines (36) and quinolones (37 and 38), were successfully incorporated into this protocol, delivering the desired products with high to excellent enantioselectivities. Notably, these transformations displayed excellent chemoselectivity towards bromides in the presence of chlorides (9, 19, 21, 23, 35), paving the way for subsequent synthetic manipulations of the products. Alkenyl bromides such as 2-bromo-1H-indene (39) and β-bromostyrene (40) turned out to be viable partners despite a slight reduction of enantioselectivity. In some cases, especially for electron-rich (hetero)aryl and alkenyl bromides, using L5 as the chiral ligand and diluting the reaction improved both the yields and enantioselectivities. Importantly, aryl bromides derived from d-glucose (41), estrone (42), (–)-menthol (43), cholesterol (44) and (+)-α-tocopherol (45) furnished the desired three-component coupling products in moderate to good yields with excellent diastereocontrol, despite the presence of several distinct C(sp3)–H bonds in the substrates, thus highlighting the chemoselectivity of the method and its applicability towards the synthesis of pharmaceutical and bioactive motifs.

The reaction and conditions are shown at the top. aStandard reaction conditions I are the same as in Fig. 2. bL5 was used as ligand, and acetone/PhCF3 (1 ml:1 ml) was used as solvent. cNiBr2·3H2O was used instead of NiBr2·DME. dTwo 40 W 390 nm Kessil lamps were used. d.r., diastereomeric ratio.
The scope of C–H precursors was explored next (Fig. 4). As expected, cycloalkanes with various ring sizes such as cyclopentane (46), cycloheptane (47), cyclooctane (48) and cyclododecane (49) were well tolerated. In the last case, because of the lack of solubility of the reagent, an e.r. of 91:9 was obtained under standard reaction conditions, which was improved to 96:4 by increasing the amount of solvent and the use of L5 as ligand. Interestingly, 2,3-dimethylbutane (50) was selectively activated on the secondary C–H bonds owing to the high bond dissociation energies of the primary C–H bonds. Remarkably, hydrocarbon derivatives bearing different electron-withdrawing groups (–Cl, –Br, –CN, –COMe, –COOMe) were successfully functionalized with excellent control of the regioselectivities and enantioselectivities (51–55). This result can be attributed to the hydridic nature of tertiary C–H bonds and the electrophilic nature of excited decatungstate65, thus providing products that are selectively functionalized distal to the electron-withdrawing moieties. Complete regioselectivity was also observed in the case of alcohols containing tertiary C–H bonds, with 2,3-dimethylbutan-2-ol affording product 56 in 81% yield and 96:4 e.r. The compatibility of diverse functional groups further emphasizes the advantages of this photo-HAT/nickel dual-catalysed strategy, as these types of alkyl radicals were inaccessible with previously developed radical precursors. This protocol is not restricted to electron-neutral, unactivated C–H systems. Ethers were regioselectively functionalized at the α-oxy position (57 and 58), which is consistent with the selectivity of decatungstate for more electron-rich C–H bonds. However, methyl tert-butyl ether, toluene and Boc-protected pyrrolidine were not able to participate in this three-component reaction, producing two-component aryl–alkyl cross-coupling products instead (Supplementary Table 9). The aforementioned reaction conditions were not suitable for the direct functionalization of hydrogen bonds in the α-position to hydroxy functions. Inspired by literature precedents25, we were delighted to find that isopropanol (2) and pentan-3-ol (59) participated in this transformation when benzophenone derivative PC I was used as the photocatalyst under modified reaction conditions. Furthermore, extraordinary regiocontrol was achieved in the presence of both tertiary and primary C–H bonds adjacent to the oxygen atom, as only the target product 60 was formed when cyclopentyl methyl ether was used as substrate. This result highlights the ability of diaryl ketone photocatalysts in site-selective activation.

We next turned our attention to expanding the scope with respect to alkenes (Fig. 4). A series of acrylates with different substituents was investigated, generating highly enantioenriched α-aryl ester products (61–66). Among them, a slight erosion in enantioselectivity was observed with less sterically hindered acrylate reactants, which was addressed by replacing ligand L1 with L5. Pleasingly, vinyl phosphonates were also suitable coupling partners under either decatungstate or diaryl ketone photocatalytic conditions. Chiral organophosphonates were obtained in good yields and excellent enantioselectivities (67 and 68). Other electron-deficient alkenes were also explored. The α,β-unsaturated ketones as well as acrylamides were viable electron acceptors, although the products (69 and 70) were obtained with moderate enantioselectivity. Tertiary acrylamides furnished a trace amount of the desired dicarbofunctionalization adducts (Supplementary Table 9). Vinylarenes, a class of activated olefins that was not compatible in previous reports49,50, were tolerated in the present system. Styrenes bearing both electron-donating groups (t-Bu) as well as electron-withdrawing groups (F, Cl, CF3) could be engaged, under slightly modified reaction conditions, to deliver the corresponding chiral 1,1-diarylalkanes with excellent enantioselectivities (71–75).
Synthetic applications
The obtained α-aryl/alkenyl carbonyls and phosphonates as well as 1,1-diarylalkanes are often found as prevalent structural motifs in pharmacologically and biologically active molecules66,67,68,69. To further demonstrate the utility of this asymmetric multicomponent reaction, transformations of the products (Fig. 5a) and synthesis of pharmaceutically relevant molecules were performed. Specifically, through acidic hydrolysis followed by esterification or amidation processes, l-tyrosine and l-tryptophan derivatives (76 and 77) were obtained with high levels of diastereocontrol, which indicated the potential of this method for rapidly accessing complex amino acid derivatives. In addition, the reduction of α-aryl ester 3 was successfully accomplished by treatment with LiAlH4, delivering alcohol 78 with excellent stereofidelity (97:3 e.r.). Because alcohols are versatile intermediates in organic synthesis, several derivatizations of 78 were also carried out. Nucleophilic substitution reactions converted 78 into the corresponding β-aryl bromide (79), β-aryl amide (80) and β-aryl thiol (81) in high yields and enantioselectivities. To illustrate its practical value further, the method was applied to a concise synthesis of two glucokinase activators. The synthetic route towards piragliatin lead compound is shown in Fig. 5b (top). The combination of commercially available cyclopentane, tert-butyl acrylate and 4-bromo-2-chloro-1-(methylsulfonyl)benzene produced the corresponding three-component cross-coupling product 83 in 65% yield and 97:3 e.r. Hydrolysis of 82 with trifluoroacetic acid followed by amidation with 2-aminopyrazine offered piragliatin lead compound 83 in 72% yield and 97:3 e.r. Compared to previous reports, this method simplifies the operation steps by using cyclopentane instead of alkyltrifluoroborate as the synthetic precursors50. Similarly, enantioenriched glucokinase activator RO28-1675 (85, 97:3 e.r.) was successfully synthesized by using the abovementioned three-step synthetic procedure (Fig. 5b, bottom).

a, Derivatization of products. b, Synthesis of piragliatin lead compound and RO28-1675. TFA, trifluoroacetic acid; DCM, dichloromethane; DCC, N,N′-dicyclohexylcarbodiimide; DMAP, 4-dimethylaminopyridine; HATU, hexafluorophosphate azabenzotriazole tetramethyl uronium; DIPEA, N,N-diisopropylethylamine; DMF, dimethylformamide; THF, tetrahydrofuran; DEAD, diethyl azodicarboxylate; DIAD, diisopropyl azodicarboxylate.
Mechanistic studies
Several experiments were designed to probe the reaction mechanism of this dual-catalytic transformation. The desired alkylarylation reaction was completely suppressed by adding 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO; 3.0 equiv.). TEMPO adducts 86 and 87 were detected by high-resolution mass spectrometry (HR-MS) in the mixture (Fig. 6a), thus demonstrating the intermediacy of an alkyl radical along the reaction pathway. Next, radical clock experiments were conducted with cyclopropyl alkene 88 under the standard reaction conditions, affording product 89 in 20% yield through a sequential radical addition, ring opening and arylation process (Fig. 6b). Product deracemization or Giese-type addition of radicals to the double bond followed by enantioselective α-arylation were ruled out based on controlled experiments (Supplementary Discussion). Subsequently, the reactivity and catalytic efficiency of presynthesized 4-CF3C6H4Ni(II)L1Br complex 90 was further investigated. Only a trace amount of 7 was detected with stoichiometric complex 90, cyclohexane and tert-butyl acrylate. One possible reason is that complex 90 is unstable in the absence of aryl bromides, and it possesses strong visible-light absorption properties54. By contrast, using 10 mol% of complex 90 instead of NiBr2·DME/L1 as catalyst in the presence of aryl bromides, product 7 was obtained in 67% yield with an identical e.r. (95:5; Fig. 6c). Taken together, these results indicate that the putative aryl-Ni(II) complex might not be a productive intermediate in the main catalytic cycle. In addition, laser flash photolysis experiments were performed to study the quenching of the excited state of TBADT (TBADT*) in the presence of increasing concentrations of cyclohexane, tert-butyl acrylate and 4-bromobenzonitrile, respectively. A clear decay of TBADT* was observed in the presence of cyclohexane following a linear Stern–Volmer behaviour (bimolecular rate constant k = 3.14 × 107 M−1 s−1). By contrast, tert-butyl acrylate and 4-bromobenzonitrile were not able to quench TBADT* (Fig. 6d). These results confirm the activation of cyclohexane by the excited state of the photocatalyst to form carbon radicals, in line with the radical trapping and clock experiments summarized in Fig. 6a,b.

a, Radical trapping experiment. b, Radical clock experiment. c, Stoichiometric and catalytic experiments with ArNi(II)L1Br complex. d, Stern–Volmer studies of TBADT with cyclohexane, tert-butyl acrylate and 4-bromobenzonitrile. e, Cyclic voltammogram studies of TBADT and L1NiBr2. f, Proposed mechanism. SET, single-electron transfer; τ, measured lifetime of excited photocatalyst with quencher; τ0, lifetime of the excited photocatalyst without quencher.
On the basis of the aforementioned mechanistic studies and precedents in the literature24, a plausible mechanism for this TBADT/nickel dual-catalysed enantioselective alkene dicarbofunctionalization can be proposed (Fig. 6f). Excited-state tetrabutylammonium decatungstate I would be formed under photoexcitation conditions, enabling the abstraction of a hydrogen atom from C(sp3)–H nucleophiles. This process generates singly reduced decatungstate II and carbon-centred radical III. Disproportionation of II provides doubly reduced decatungstate IV and regenerates ground-state TBADT. In parallel, radical addition of III to an olefin would form adduct V, which can be captured by Ni(0) species VI to furnish alkyl-Ni(I) intermediate VII. Subsequent oxidative addition of alkenyl bromide to Ni(I) species VII generates alkyl-Ni(III)-aryl intermediate VIII. Reductive elimination produces the desired dicarbofunctionalization product as well as Ni(I) species IX. Both catalytic cycles converge to completion through final single-electron transfer between this Ni(I) species and doubly reduced TBADT IV to regenerate the active Ni(0) catalyst VI and singly reduced TBADT II. This process is supported by cyclic voltammetry studies (Fig. 6e), as the reductive potential of [W10O32]5–/[W10O32]6– (E1/2 = –1.85 V versus Fc/Fc+ in MeCN, reductive peak observed at –1.93 V) is more negative than that of NiI/Ni0 (E1/2 = –1.74 V versus Fc/Fc+ in MeCN, reductive peak observed at –1.82 V). A NiI/NiII/NiIII catalytic sequence is also possible (Fig. 6f, grey). Alternatively, Ni(I) species VII could be obtained through the combination of Ni(I) species IX with radical adduct V, followed by the subsequent single-electron reduction of alkyl-Ni(II) intermediate X.
Conclusions
In summary, the combination of photoredox-mediated HAT with a nickel-catalysed radical relay has enabled a highly enantioselective three-component difunctionalization of alkenes. Unactivated hydrocarbons can be directly used as coupling agents without the requirement of pre-activated radical precursors, making this strategy more attractive for step-economical and atom-economical synthesis. Site-selective activation and the conversion of alkanes, ethers and alcohols were achieved by employing decatungstate or diaryl ketone photocatalysts. The method exhibits mild conditions, broad substrate scope and excellent regioselectivity, chemoselectivity and enantioselectivity, providing a versatile synthesis of value-added enantioenriched α-aryl/alkenyl carbonyls/phosphonates and 1,1-diarylalkanes. The synthetic potential of this method has been demonstrated by derivatization of the products and the synthesis of medicinally relevant molecules. We believe that the lessons obtained here will inspire the development of more asymmetric multicomponent reactions enabled by C(sp3)–H activation in the future.
Methods
Method A
In a nitrogen-filled glove box, a reaction vial (5 ml) equipped with a stirring bar was charged with TBADT (0.004 mmol, 2 mol%), NiBr2·DME (0.02 mmol, 10 mol%), (4 S,4′S)-4,4′-di((S)-sec-butyl)-1,1′-bis(3-(tert-butyl)phenyl)-4,4′,5,5′-tetrahydro-1H,1′H-2,2′-biimidazole (L1; 0.03 mmol, 15 mol%) or (4 S,4′S)-4,4′-di((S)-sec-butyl)-1,1′-bis(3,5-di-tert-butylphenyl)-4,4′,5,5′-tetrahydro-1H,1′H-2,2′-biimidazole (L5; 0.03 mmol, 15 mol%), anhydrous K3PO4 (0.4 mmol, 2 equiv.), aryl bromide (0.2 mmol, 1 equiv.), C–H radical precursor (2 mmol, 10 equiv.), alkene (0.6 mmol, 3 equiv.), dry acetone (0.5 ml, 1.0 ml when L5 was used) and dry α,α,α-trifluorotoluene (0.5 ml, 1.0 ml when L5 was used). The vial was sealed and removed from the glove box. The reaction mixture was prestirred for 20 min, then irradiated with a Kessil PR160 390 nm lamp at 5 °C with a distance of ~3 cm from the surface of the reaction vial. After 18 h of irradiation, the resulting mixture was passed through a pipette plug of Celite and silica gel and eluted with EtOAc. After concentration under reduced pressure, the crude mixture was purified by chromatography on silica gel with hexane/EtOAc mixtures to give the corresponding products.
Method B
In a nitrogen-filled glove box, a reaction vial (5 ml) equipped with a stirring bar was charged with (4-methoxyphenyl)(4-(trifluoromethyl)phenyl)methanone (PC I; 0.04 mmol, 20 mol%), NiBr2·DME (0.02 mmol, 10 mol%), L5 (0.03 mmol, 15 mol%), anhydrous Na2CO3 (0.4 mmol, 2 equiv.), aryl bromide (0.2 mmol, 1 equiv.), C–H radical precursor (2 mmol, 10 equiv.), alkene (0.6 mmol, 3 equiv.), dry acetone (1.0 ml) and dry α,α,α-trifluorotoluene (1.0 ml). The vial was sealed and removed from the glove box. The reaction mixture was prestirred for 20 min, then irradiated with a Kessil PR160 390 nm lamp at 5 °C with a distance of ~3 cm from the surface of the reaction vial. After 18 h of irradiation, the resulting mixture was passed through a pipette plug of Celite and silica gel and eluted with EtOAc. After concentration under reduced pressure, the crude mixture was purified by chromatography on silica gel with hexane/EtOAc mixtures to give the corresponding products.
Data availability
The X-ray crystallographic coordinates for compound 1 reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2268422. These data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/data_request/cif. The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information. Data are available from the corresponding author upon request.
References
Labinger, J. A. & Bercaw, J. E. Understanding and exploiting C–H bond activation. Nature 417, 507–514 (2002).
Zhang, C., Li, Z. L., Gu, Q. S. & Liu, X. Y. Catalytic enantioselective C(sp3)–H functionalization involving radical intermediates. Nat. Commun. 12, 475 (2021).
Saint-Denis, T. G. et al. Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts. Science 359, eaao4798 (2018).
Olden, D. L., Suh, S. E. & Stahl, S. S. Radical C(sp3)–H functionalization and cross-coupling reactions. Nat. Rev. Chem. 6, 405–427 (2022).
Capaldo, L., Ravelli, D. & Fagnoni, M. Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C–H bonds elaboration. Chem. Rev. 122, 1875–1924 (2022).
Cao, H., Tang, X., Tang, H., Yuan, Y. & Wu, J. Photoinduced intermolecular hydrogen atom transfer reactions in organic synthesis. Chem. Catal. 1, 523–598 (2021).
Capaldo, L. & Ravelli, D. Hydrogen atom transfer (HAT): a versatile strategy for substrate activation in photocatalyzed organic synthesis. Eur. J. Org. Chem. 2017, 2056–2071 (2017).
Ravelli, D., Fagnoni, M., Fukuyama, T., Nishikawa, T. & Ryu, I. Site-selective C–H functionalization by decatungstate anion photocatalysis: synergistic control by polar and steric effects expands the reaction scope. ACS Catal. 8, 701–713 (2017).
Tzirakis, M. D., Lykakis, I. N. & Orfanopoulos, M. Decatungstate as an efficient photocatalyst in organic chemistry. Chem. Soc. Rev. 38, 2609–2621 (2009).
West, J. G., Huang, D. & Sorensen, E. J. Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 6, 10093 (2015).
Cao, H. et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 11, 1956 (2020).
Perry, I. B. et al. Direct arylation of strong aliphatic C–H bonds. Nature 560, 70–75 (2018).
Shen, Y., Gu, Y. & Martin, R. sp3 C–H arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J. Am. Chem. Soc. 140, 12200–12209 (2018).
Sarver, P. J. et al. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem. 12, 459–467 (2020).
Murphy, J. J., Bastida, D., Paria, S., Fagnoni, M. & Melchiorre, P. Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals. Nature 532, 218–222 (2016).
Laudadio, G. et al. C(sp3)–H functionalizations of light hydrocarbons using decatungstate photocatalysis in flow. Science 369, 92–96 (2020).
Laudadio, G. et al. Selective C(sp3)–H aerobic oxidation enabled by decatungstate photocatalysis in flow. Angew. Chem. Int. Ed. 57, 4078–4082 (2018).
Schultz, D. M. et al. Oxyfunctionalization of the remote C–H bonds of aliphatic amines by decatungstate photocatalysis. Angew. Chem. Int. Ed. 56, 15274–15278 (2017).
Ryu, I. et al. Efficient C–H/C–N and C–H/C–CO–N conversion via decatungstate-photoinduced alkylation of diisopropyl azodicarboxylate. Org. Lett. 15, 2554–2557 (2013).
Wan, T. et al. Decatungstate-mediated C(sp3)–H heteroarylation via radical–polar crossover in batch and flow. Angew. Chem. Int. Ed. 60, 17893–17897 (2021).
Halperin, S. D., Fan, H., Chang, S., Martin, R. E. & Britton, R. A convenient photocatalytic fluorination of unactivated C–H bonds. Angew. Chem. Int. Ed. 53, 4690–4693 (2014).
Xia, J. B., Zhu, C. & Chen, C. Visible light-promoted metal-free C–H activation: diarylketone-catalyzed selective benzylic mono- and difluorination. J. Am. Chem. Soc. 135, 17494–17500 (2013).
Sarver, P. J., Bissonnette, N. B. & MacMillan, D. W. C. Decatungstate-catalyzed C(sp3)–H sulfinylation: rapid access to diverse organosulfur functionality. J. Am. Chem. Soc. 143, 9737–9743 (2021).
Xu, S., Chen, H., Zhou, Z. & Kong, W. Three-component alkene difunctionalization by direct and selective activation of aliphatic C–H bonds. Angew. Chem. Int. Ed. 60, 7405–7411 (2021).
Campbell, M. W., Yuan, M., Polites, V. C., Gutierrez, O. & Molander, G. A. Photochemical C–H activation enables nickel-catalyzed olefin dicarbofunctionalization. J. Am. Chem. Soc. 143, 3901–3910 (2021).
Lu, F. D., He, G. F., Lu, L. Q. & Xiao, W. J. Metallaphotoredox catalysis for multicomponent coupling reactions. Green Chem. 23, 5379–5393 (2021).
Wickham, L. M. & Giri, R. Transition metal (Ni, Cu, Pd)-catalyzed alkene dicarbofunctionalization reactions. Acc. Chem. Res. 54, 3415–3437 (2021).
Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287–4296 (2020).
Qi, X. & Diao, T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal. 10, 8542–8556 (2020).
Badir, S. O. & Molander, G. A. Developments in photoredox/nickel dual-catalyzed 1,2-difunctionalizations. Chem 6, 1327–1339 (2020).
Zhu, C., Yue, H., Chu, L. & Rueping, M. Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chem. Sci. 11, 4051–4064 (2020).
Campbell, M. W., Compton, J. S., Kelly, C. B. & Molander, G. A. Three-component olefin dicarbofunctionalization enabled by nickel/photoredox dual catalysis. J. Am. Chem. Soc. 141, 20069–20078 (2019).
Garcia-Dominguez, A., Mondal, R. & Nevado, C. Dual photoredox/nickel-catalyzed three-component carbofunctionalization of alkenes. Angew. Chem. Int. Ed. 58, 12286–12290 (2019).
Guo, L., Tu, H. Y., Zhu, S. & Chu, L. Selective, intermolecular alkylarylation of alkenes via photoredox/nickel dual catalysis. Org. Lett. 21, 4771–4776 (2019).
Mega, R. S., Duong, V. K., Noble, A. & Aggarwal, V. K. Decarboxylative conjunctive cross-coupling of vinyl boronic esters using metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 4375–4379 (2020).
Zheng, S. et al. Selective 1,2-aryl-aminoalkylation of alkenes enabled by metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 17910–17916 (2020).
Li, Z. L., Fang, G. C., Gu, Q. S. & Liu, X. Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32–48 (2020).
Fu, L., Zhou, S., Wan, X., Chen, P. & Liu, G. Enantioselective trifluoromethylalkynylation of alkenes via copper-catalyzed radical relay. J. Am. Chem. Soc. 140, 10965–10969 (2018).
Lin, J. S. et al. Cu/chiral phosphoric acid-catalyzed asymmetric three-component radical-initiated 1,2-dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 141, 1074–1083 (2019).
Wang, F. et al. Enantioselective copper-catalyzed intermolecular cyanotrifluoromethylation of alkenes via radical process. J. Am. Chem. Soc. 138, 15547–15550 (2016).
Wang, P. Z. et al. Asymmetric three-component olefin dicarbofunctionalization enabled by photoredox and copper dual catalysis. Nat. Commun. 12, 1815 (2021).
Wu, L., Wang, F., Chen, P. & Liu, G. Enantioselective construction of quaternary all-carbon centers via copper-catalyzed arylation of tertiary carbon-centered radicals. J. Am. Chem. Soc. 141, 1887–1892 (2019).
Wu, L. et al. Asymmetric Cu-catalyzed intermolecular trifluoromethylarylation of styrenes: enantioselective arylation of benzylic radicals. J. Am. Chem. Soc. 139, 2904–2907 (2017).
Zhu, S., Zhao, X., Li, H. & Chu, L. Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel. Chem. Soc. Rev. 50, 10836–10856 (2021).
Chierchia, M., Xu, P., Lovinger, G. J. & Morken, J. P. Enantioselective radical addition/cross-coupling of organozinc reagents, alkyl iodides, and alkenyl boron reagents. Angew. Chem. Int. Ed. 58, 14245–14249 (2019).
Tu, H. Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604–9611 (2020).
Wei, X., Shu, W., Garcia-Dominguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515–13522 (2020).
Wang, F., Pan, S., Zhu, S. & Chu, L. Selective three-component reductive alkylalkenylation of unbiased alkenes via carbonyl-directed nickel catalysis. ACS Catal. 12, 9779–9789 (2022).
Qian, P. et al. Catalytic enantioselective reductive domino alkyl arylation of acrylates via nickel/photoredox catalysis. Nat. Commun. 12, 6613 (2021).
Guo, L. et al. General method for enantioselective three-component carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. 142, 20390–20399 (2020).
Li, X. et al. Three-component enantioselective alkenylation of organophosphonates via nickel metallaphotoredox catalysis. Chem 9, 154–169 (2023).
Du, X. Y., Cheng-Sánchez, I. & Nevado, C. Dual nickel/photoredox-catalyzed asymmetric carbosulfonylation of alkenes. J. Am. Chem. Soc. 145, 12532–12540 (2023).
Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C–H arylation via photoredox and nickel dual catalysis. Nat. Commun. 10, 3549 (2019).
Rand, A. W. et al. Dual catalytic platform for enabling sp3 α-C–H arylation and alkylation of benzamides. ACS Catal. 10, 4671–4676 (2020).
Shu, X., Huan, L., Huang, Q. & Huo, H. Direct enantioselective C(sp3)–H acylation for the synthesis of α-amino ketones. J. Am. Chem. Soc. 142, 19058–19064 (2020).
Cheng, X. et al. Stereo- and enantioselective benzylic C–H alkenylation via photoredox/nickel dual catalysis. ACS Catal. 11, 11059–11065 (2021).
Xu, J., Li, Z., Xu, Y., Shu, X. & Huo, H. Stereodivergent synthesis of both Z- and E-alkenes by photoinduced, Ni-catalyzed enantioselective C(sp3)–H alkenylation. ACS Catal. 11, 13567–13574 (2021).
Shu, X., Zhong, D., Lin, Y., Qin, X. & Huo, H. Modular access to chiral α-(hetero)aryl amines via Ni/photoredox-catalyzed enantioselective cross-coupling. J. Am. Chem. Soc. 144, 8797–8806 (2022).
Shu, X., Zhong, D., Huang, Q., Huan, L. & Huo, H. Site- and enantioselective cross-coupling of saturated N-heterocycles with carboxylic acids by cooperative Ni/photoredox catalysis. Nat. Commun. 14, 125 (2023).
Xu, S. et al. Enantioselective C(sp3)–H functionalization of oxacycles via photo-HAT/nickel dual catalysis. J. Am. Chem. Soc. 145, 5231–5241 (2023).
Zhang, W. et al. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016).
Li, J. et al. Site-specific allylic C–H bond functionalization with a copper-bound N-centred radical. Nature 574, 516–521 (2019).
Li, Y., Lei, M. & Gong, L. Photocatalytic regio- and stereoselective C(sp3)–H functionalization of benzylic and allylic hydrocarbons as well as unactivated alkanes. Nat. Catal. 2, 1016–1026 (2019).
Xu, P., Fan, W., Chen, P. & Liu, G. Enantioselective radical trifluoromethylation of benzylic C–H bonds via cooperative photoredox and copper catalysis. J. Am. Chem. Soc. 144, 13468–13474 (2022).
Waele, V. D., Poizat, O., Fagnoni, M., Bagno, A. & Ravelli, D. Unraveling the key features of the reactive state of decatungstate anion in hydrogen atom transfer (HAT) photocatalysis. ACS Catal. 6, 7174–7182 (2016).
Bhutani, P. et al. FDA approved drugs from 2015–June 2020: a perspective. J. Med. Chem. 64, 2339–2381 (2021).
Lamberth, C. & Dinges, J. (eds) Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals (Wiley-VCH, 2016).
Horsman, G. P. & Zechel, D. L. Phosphonate biochemistry. Chem. Rev. 117, 5704–5783 (2017).
Ameen, D. & Snape, T. J. Chiral 1,1-diaryl compounds as important pharmacophores. MedChemComm 4, 893–907 (2013).
Acknowledgements
We thank O. Blacque for the X-ray diffraction analysis of 1 (Cambridge Crystallographic Data Centre no. 2268422). We thank P. Hamm, J. Helbing and K. Oppelt for laser flash photolysis experiments. We thank N. Cramer and G. Zhang for HPLC testing of 72, 74 and 75. This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation. We also acknowledge the Swiss National Science Foundation (SNF es 200021_184986/1 to C.N.) for financial support.
Funding
Open access funding provided by University of Zurich.
Author information
Authors and Affiliations
Contributions
C.N. and X.H. conceived the project. X.H. and I.C.-S. performed the experiments. C.N., X.H., I.C.-S., W.K. and G.A.M. analysed the data and co-wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental procedures, characterization data, NMR spectra, HPLC traces and crystallographic data.
Supplementary Data
Crystallographic data for compound 1; CCDC reference 2268422.
Supplementary Data
CheckCIF report for compound 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, X., Cheng-Sánchez, I., Kong, W. et al. Nickel-catalysed enantioselective alkene dicarbofunctionalization enabled by photochemical aliphatic C–H bond activation. Nat Catal (2024). https://doi.org/10.1038/s41929-024-01153-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41929-024-01153-0
