
Abstract
BRAF mutations are a significant driver of disease in pediatric low-grade glioma, but the implications of BRAF alterations on the clinical course and treatment response in adult glioma remain unclear. Here, we characterize a multi-institutional cohort of more than 300 patients (>200 adults) with BRAF-mutated glioma using clinical, pathological/molecular, and outcome data. We observed that adult and pediatric BRAF-mutant gliomas harbor distinct clinical and molecular features, with a higher prevalence of BRAFV600E (Class I) and BRAF fusions in pediatric tumors. BRAFV600E alterations were associated with improved survival in adults with glioma overall, though not in glioblastoma. Other genomic alterations observed within functional classes were consistent with the putative roles of those BRAF mutation classes in glioma pathogenesis. In our adult cohort, BRAFV600E alterations conferred sensitivity to targeted therapies. Overall, this large cohort of BRAF-altered adult gliomas demonstrates a broad range of molecular alterations with implications for treatment sensitivity and survival.
Similar content being viewed by others


Introduction
BRAF (v-raf murine viral oncogene homolog B1), a member of the RAF family of serine/threonine protein kinases that signal to MEK-ERK kinases, has been identified as an oncogene driver in multiple cancers. BRAF mutations have been classified into three categories by their functional impact on BRAF kinase activity, dimerization, and RAS dependency1,2. The most common BRAF alteration (Class I), p.V600E, functions as a monomer to promote ERK signaling. Less common Class II alterations facilitate ERK signaling through increased homo- and hetero-dimerization of BRAF and do not require upstream activation by RAS or receptor tyrosine kinases (RTKs)3. Class III BRAF alterations are functionally inactivating alterations that increase the proclivity of BRAF to dimerize with CRAF, thereby increasing downstream ERK signaling4. Class I alterations are targetable with FDA-approved small molecule inhibitors, while novel drugs against other BRAF alterations and pathway components are under development5.
The role of BRAF alterations has been a topic of focused interest in pediatric glioma given the high prevalence of p.V600E (Class I) and BRAF-KIAA1549 fusions (Class II)6,7, with early evidence of success by BRAF-targeted therapy in this group8,9,10. BRAF alterations are also observed in adults with low- (5%) and high-grade glioma (3%)5, prompting considerable interest in targeting oncogenic BRAF alterations in adults11,12. To date, our understanding of the spectrum of BRAF alterations and their implications for disease trajectory and treatment response in adults remains limited.
Recent efforts to elucidate the co-occurrence of BRAF alterations with other genomic mutations and patient outcomes in adult gliomas are limited by case numbers or a particular focus on specific alterations, such as canonical BRAFV600E mutations and BRAF amplifications13,14,15,16. Studies have not investigated the range of BRAF mutations, co-occurring alterations, and their clinical implications across a large set of sequenced genes, nor have there been robust comparisons of BRAF alteration types between adult and pediatric gliomas.
In this study, we retrospectively characterize a large multi-institutional cohort of adults with BRAF-mutated gliomas. We identify the clinical phenotypes, genomic signatures, and molecular features associated with different functional classes to assess prognostic implications and guide therapeutic approaches.
Results
Patient cohort
We obtained clinical and molecular data from 296 patients with BRAF-altered glioma (151 males, 145 females), including 206 adults (median 43 years, range 18–85 years) and 90 children (age <18 years, median 10 years, range 0–17 years). Subsets of this cohort had molecular (n = 267) and clinical outcomes (n = 216) data, with the majority of patients (n = 187) having both (Fig. 1a). Most samples were obtained at initial diagnosis (n = 220), with 56 from a follow-up resection or at an unknown time (n = 20).

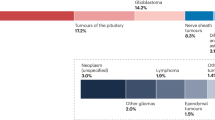
a Study schematic demonstrating the source of cases and six cohorts into which BRAF alterations were categorized based on their known or presumed function. b Proportion of tumors with key pathological features among glioma samples from 206 adults and 90 pediatric patients. c Composition and clinical features of adult glioma classes by BRAF alteration including age at diagnosis, clinical histo-pathologic diagnosis, tumor grade, and overall survival (when known).
The spectrum of histologic grades, pathology, and specific BRAF alterations in adults contrasted with that in children (Fig. 1b and Table 1). The most common histopathologic diagnosis in adults was glioblastoma (n = 108, 36%) (Table 1 and Fig. 1b, c). By contrast, low-grade gliomas, especially pilocytic astrocytomas, predominated in pediatric BRAF-altered cases (p = 0.0003). BRAF fusions and Class I mutations were most prevalent in children, while adult gliomas harbored a broad range of BRAF alterations, including oncogenic Class II/III alterations, copy number alterations, and otherwise unclassified alterations, all of which were rare in pediatric glioma.
Landscape of BRAF alterations in adult glioma
We explored the distribution of BRAF alterations across age and histopathologic groups, along with associations with other common alterations (IDH1/2, EGFR, TERT, ATRX, H3K27M, CDKN2A/B, NF1, KRAS, HRAS). While the most common SNV in BRAF was at p.V600E in both adult and pediatric gliomas, alterations at other sites known to affect BRAF dimerization or increase ERK signaling (Class II/III) were more common in adult samples (Fig. 2a, b). In addition to the previously described Class II and III alterations3,4, we observed a small number of alterations in close proximity to those nucleotides which likely occur in functional domains but have not yet been validated (within kinase domain: Q461E, I463M, V471I, G474R, V482G, L584I, S614P, S614P; within receptor-binding domain: R188K, G209S). Other alterations distant from the kinase domain may be functionally insignificant and incidental. For this analysis, samples were included in the BRAF Class II and III cohorts only if the SNV was previously shown to be ERK-activating1.

Lollipop plot depicting the location of BRAF single-nucleotide variants (SNVs) identified in the a adult or b pediatric glioma cohort, with color denoting alteration class. c Co-mutation plot grouped by BRAF alteration class for all samples with genomic profiling data.
Approximately half of adults in our cohort had Class I BRAF alterations (n = 103/206; median age at diagnosis 39 years, range 18–86 years; 57% female). GBM was the most common histopathologic diagnosis (51%, n = 52), followed by low-grade glioma (22%), and PXA (18%), among others (Table 1). Overall survival across adults with Class I alterations was 165 months (Fig. 1c). In total, 2% (n = 2) had mutations in IDH1/2, 28% (n = 29) had loss of CDKN2A/B, 3.9% (n = 4) had alterations in ATRX, none had alterations in H3K27 or NF1. We found 24% of BRAFV600E-mutant GBMs (n = 10/41) had mutations in the TERT promoter, 22% (n = 9) had chromosome 7p gain and 10q loss, and none had EGFR amplification. A subset of gliomas across all ages with Class I BRAF mutations harbored no other alterations, including a subset histologically defined as GBMs (Fig. 2c).
Class II alterations (3.9%; median age 44 years, range 28–76 years; Figs. 1c and 2c) were seen in eight adult and no pediatric patients. The alterations occurred at three amino acids (p.L597R, p.G469R/V/A, and p.K601E) previously described in melanoma3. Histopathology was predominantly GBM, though there were four grade 2 gliomas as well—one diffuse astrocytoma (IDH-WT), one oligodendroglioma (IDH-mutant, 1p/19q co-deleted), and two astrocytomas, IDH-mutant. Median survival was 20 months.
While Class II alterations are not dependent on RAS activation, we did observe NF1 alterations in three specimens (38%). Of note, three specimens had concurrent IDH1/2 mutations, two of which were known oncogenic alterations (Fig. 2).
Nine (4.3%; median age 55 years, range 28–76 years) patients in our adult cohort had Class III alterations (Fig. 1c and Table 1). These included p.G466R/E, p.G469E, p.N581S, p.D594G/N, and p.G596D. All but one tumor with Class III BRAF alterations were glioblastomas, while the remaining one was a grade 3 IDH-mutant astrocytoma. Median survival was 47 months. One pediatric patient with a grade 3 HGG harbored a Class III alteration.
Given their dependence on RAS activity, Class III alterations commonly co-occur with RTK alterations, NF1 loss, or RAS-activating alterations. We observed mutations in RTK, RAS, or ERK pathway components in all adults with Class III altered gliomas (n = 9). Specifically, seven had NF1 alterations, two had activating EGFR alterations, and one had an alteration in MET. Additionally, two had TERT-promoter mutations, four had CDKN2A/B losses, and one had an IDH1/2 alteration (Fig. 2).
BRAF rearrangements are common in pediatric LGG, particularly pilocytic astrocytoma, and linked with favorable survival7. Twenty adult patients (9.7%; median age 42 years; range 18–76 years) had tumors harboring BRAF rearrangements (Fig. 1c and Table 1). Of these, 10 (50%) were PA, 6 (30%) were GBM, 2 (10%) were low-grade glioma, and 2 (10%) were astrocytoma, IDH-mutant (one grade 2 and one grade 4). The most common BRAF rearrangement partner was KIAA1549 (n = 11) (Fig. 1b). Two BRAF-rearranged gliomas had TERT promoter mutations and five had loss of CDKN2A/B, all of which were grade 4. Median survival in the entire cohort was 71 months; 33 months in patients with GBM and not reached in those with PA.
In the pediatric cohort, BRAF rearrangements dominated (n = 55; 61%). The majority of rearrangements were KIAA1549-BRAF rearrangements (n = 47; 85%), with only one tumor each harboring BCAS1, CCDC6, GIT2, or PTPRZ11 as a fusion partner (Fig. 1b). Clinically, all but two (1.8%) patients were diagnosed with a pilocytic astrocytoma, with the remaining being grade 2 PXA or LGG (Supplementary Fig. 1).
Twenty-seven adults had tumors with BRAF copy number gains. Age at diagnosis was bimodal with a peak at 35 years and second peak at 75 years. The majority of patients (70%, n = 19) had glioblastoma, while 9 tumors (33%) had IDH mutations. Median survival in this cohort was 39 months. Only one pediatric patient had a BRAF gain; histopathologic diagnosis was GBM, IDH-WT and clinical deterioration was rapid.
Tumors with unclassified BRAF alterations comprised 19% of the adult cohort (n = 39) and 6% of pediatric cases (n = 5) (Figs. 1b and 2c). In adults, median age at diagnosis was 49 years (range 21–78). Seventeen (44%) were female. The majority of alterations in this cohort were SNVs (n = 30, 77%), with 4 intragenic rearrangements, 2 having focal loss of BRAF, and 3 damaging mutations (Fig. 2c). Median overall survival in this cohort was 42 months.
Interestingly, a subset of unclassified adult tumors (n = 13, 6.3%) had increased tumor mutation burden across the genome, consistent with a hypermutated phenotype (Fig. 2c). Three hypermutated tumors had known pathogenic IDH mutations, while four had IDH mutations of unknown significance. Nine were newly diagnosed gliomas and 4 were recurrent tumors. Of four patients with detailed clinical treatment data, all received temozolomide prior to the surgical specimen for which NGS was obtained.
Association of molecular alterations with BRAF class
We next evaluated associations between different BRAF alteration classes and other molecular characteristics. We performed unsupervised clustering using genomic data, which identified 3 clusters. Gliomas with BRAF gains (Cluster 2) were more closely associated with one another than gliomas with BRAF Class I–III mutations or fusions (Fig. 3a and Supplementary Table 1). A small cluster (Cluster 3) of primarily high-grade gliomas contained mostly unclassified BRAF alterations. Gliomas with BRAF gains commonly had concomitant gains in MET, SMO, EZH2, and CDK6 (p < 0.001 each; Supplementary Table 2), along with other genes on 7q, which may reflect larger chromosomal duplications (Fig. 3b). Interestingly, HRAS, MYC, and TP53 alterations were also enriched in the gains cohort (p < 0.00001).

a Logistic PCA plot with k-means clustering of BRAF-altered gliomas based on genomic alterations. b Gene alterations significantly enriched or depleted across BRAF-altered glioma classes, shaded by chi-square residuals on post hoc analysis where red and blue signify positive and negative relationships, respectively. c Top co-occurring gene mutations by BRAF alteration class, colored bar thickness represents number of cases. d Structural DNA rearrangements in 20 adult gliomas and 55 pediatric gliomas. Lines connect the chromosomal locations of BRAF and fusion gene partners, line thickness is proportional to rearrangement frequency in study cohort. e Differentially expressed genes in BRAF Class I tumors versus other BRAF-altered gliomas in transcript sequence data from TCGA samples. f Univariate Cox proportional hazards analysis evaluating associations between molecular alterations and overall survival in 187 adult glioma patients. Hazard ratios (diamonds) and 95% confidence intervals (horizontal lines) are depicted.
In BRAFV600E-altered gliomas, we found no significant co-occurring oncogenes when compared with other alteration classes. However, Class I alterations were negatively correlated with alterations in NF1, EGFR, TP53, and MET (Fig. 3b, c). As anticipated, concurrent alterations in RAS pathway components were rarely observed (Fig. 2c).
Several other correlations were observed between classes, some anticipated based on known activity (Fig. 3b, c and Supplementary Table 2). Tumors with Class II and III alterations were enriched for NF1 alterations (Fig. 3a; p < 0.00001). EGFR and FGFR4 alterations were associated with unclassified BRAF alterations after excluding hypermutated samples (p = 0.0003). Notably, CDKN2A/B loss, ATRX, and PTEN mutations were observed across all groups.
BRAF rearrangements were not associated with other molecular alterations in our panel. Within the BRAF rearranged cases, non-KIAA1549 rearrangements were associated with higher pathologic grade. Of note, all rearrangements in adult glioma were intrachromosomal rearrangements on the long arm of chromosome 7, while in pediatric glioma several interchromosomal rearrangements were observed (Fig. 3d).
To further explore differences between tumors with distinct BRAF alterations, we analyzed RNA sequencing data from 18 TCGA samples in our cohort. These tumors harbored Class I alterations (n = 4), BRAF gains (n = 10), or unclassified alterations (n = 4). Compared to other BRAF-altered gliomas, tumors with Class I alterations had elevated expression of several well-known tumor microenvironment marker genes (Fig. 3e). Notably, gliomas with Class I alterations were enriched for transcripts associated with MEK functional activation (e.g., ETV4, LZTS1), which can also be markers of MEK inhibitor sensitivity17,18. They also demonstrated increased expression of EREG (an EGFR ligand) and the EPHA2 receptor, which are associated with resistance to treatment with EGFR or BRAF inhibitors19,20. This finding is consistent with recent observations that BRAF-mutant GBM have distinct expression profiles compared with non-BRAF-mutant GBM16. Meanwhile, tumors with BRAF gains were enriched for genes associated with transcriptional regulation compared to other samples (Supplementary Fig. 2).
Impact of BRAF class on clinical outcome
We evaluated the impact of clinical and molecular features on outcomes in this retrospective cohort of 187 patients (127 adults, 60 children) with combined data. Diagnosis of pilocytic astrocytoma, oligodendroglioma, and younger age at diagnosis were associated with more favorable prognoses, while age >50 years, GBM pathology, IDH-WT status, and higher tumor grade were associated with inferior prognoses (p < 0.001) (Fig. 3f).
Molecular features such as CDKN2A/B loss, PTEN, and TERT promoter mutations were associated with inferior prognoses (p < 0.001). Other alterations associated with worse prognosis across pediatric and adult patients included alterations in AGK, MET, CREBBP, STAG2, PARK2, CCND2, DNMT3A, PRDM1, and IKZF1 (p < 0.05; Fig. 3f). Some alterations associated with worse prognosis (PTEN, EGFR, NF1) correlated with increased age (p < 0.0005; Supplementary Table 2). The only alteration associated with improved survival was KIAA1549, as expected given its association with pilocytic astrocytoma.
Gliomas with distinct BRAF alteration types exhibited varying survival (Fig. 1c). We found that overall survival was significantly prolonged in adults with Class I alterations compared to other alterations (p = 0.032; Fig. 4a). This appears to be partially driven by grade, as no survival difference was observed between GBM patients with Class I BRAF alterations and those with non-Class I alterations (p = 0.67, Fig. 4b), with median survival of 22 months across patients with GBM. This analysis did not control for the type or number of lines of therapy.

Survival of adults with BRAF V600E alterations (blue) versus all other alterations (gray) for a all glioma or b glioblastoma only. c Overall survival for adult and pediatric patients with gliomas harboring Class I alterations or BRAF rearrangements separated by age and alteration type. d Overall survival of patients with glioblastoma by age categories. e Survival in adults with BRAF rearrangements separated by tumor grade. f Overall survival of adults with high-grade glioma treated (blue) or not treated (red) with targeted therapy.
Notably, our cohort demonstrated a significant difference in survival between pediatric and adult patients with either BRAF Class I alterations or rearrangements (p = 0.0012; Fig. 4c), underscoring the large difference in prognosis between adult and pediatric tumors, even with the same molecular alteration. Younger age remained associated with improved survival in 71 adult GBMs with survival data (p < 0.0001, Fig. 4d).
Additionally, we observed similar survival between adult patients with Class I alterations and rearrangements (Fig. 4c). When adults with rearrangements were separated by grade, there was a survival difference between grade 1 and grade 4 gliomas, suggesting grade serves as a more accurate prognostic indicator than the presence of a BRAF rearrangement alone (Fig. 4e). In adults with grade 3–4 astrocytoma, BRAF/MEK-targeted therapy was associated with improved overall survival (Fig. 4f) compared with grade 3–4 patients who did not receive targeted therapy, suggesting targeted therapy has the ability to impact disease trajectory.
Effect of targeted therapy in adults with BRAF mutant glioma
Thirteen adults with recurrent gliomas received BRAF/MEK-targeted therapy (Fig. 5a), two of whom have been previously described21,22. Eleven had Class I mutations and 2 had non-Class I alterations. Median age at diagnosis was 26 years and 38% (n = 5) were female. Targeted therapy consisted of BRAF inhibition (vemurafenib, n = 3; vemurafenib followed by dabrafenib, n = 1), MEK inhibition (trametinib, n = 1), or a combination (dabrafenib and trametinib, n = 8). BRAF and/or MEK inhibition was first-line therapy in two patients. However, the majority of patients had BRAF and/or MEK inhibition either initiated as second-line therapy following radiation with or without concurrent temozolomide (n = 5, 38%), or initiated after multiple lines of therapy (n = 6, 46%). Six patients had stable disease for four or more months on targeted therapy. Median time to progression while on targeted therapy was 5.0 months. Overall survival for all patients receiving targeted therapy was 165 months and 53 months for patients with GBM. Four patients (31%) in the cohort were continuing targeted therapy at the time of last follow-up. Reasons for discontinuation of therapy were treatment-related side effects (n = 4), radiological evidence of progression (n = 2), death (n = 2), or unknown (n = 1).

a Swimmers plot depicting clinical course of patients indicating time on BRAF-targeted therapy (red), radiation alone or radiation combined with temozolomide (blue), temozolomide treatment alone (green), other anti-neoplastic treatment (gray), or observation (dashed line). Time of events are noted for progression (open circle), remission (open diamond), death (black diamond), or continued treatment and/or observation (arrow). All patients are centered on the time they initiated targeted therapy, with lines to the left of midline depicting clinical course prior to targeted therapy. All patients had a BRAF V600E mutation on molecular analysis, with the exceptions of patients “GBM-4” and “PA-1”, whose gliomas had a BRAF Class III (N581S) mutation and KIAA1549-BRAF fusion, respectively. b Graphic depiction of clinical course in a patient with epithelioid GBM treated with multiple lines of BRAF-targeted therapy.
The complexities of BRAF targeted therapy are highlighted by one patient whose case has been partially described previously (Fig. 5b)21. She was diagnosed at 23 years of age with epithelioid glioblastoma and treated with radiation and concurrent temozolomide, followed by 12 cycles of adjuvant temozolomide. Her cancer recurred 5 years after diagnosis, at which time she underwent repeat resection with carmustine wafer placement. Two years later, she had a second recurrence and started dabrafenib plus trametinib for 44 months before discontinuing therapy due to neutropenia. She was monitored clinically with no evidence of recurrence for 14 months, before another radiographic recurrence prompted repeat resection, carmustine wafer placement, and initiation of encorafenib with binimetinib. She remained on therapy for 6 months before discontinuation due to colitis and then remained disease-free for another 6 months before recurrence.
Discussion
While BRAF alterations are significant and potentially targetable in glioma, the frequency, spectrum, and clinical implications of BRAF alterations in adult glioma have not been previously characterized due to their rarity. In comparison to prior studies, this multi-institutional cohort has improved coverage of clinical outcomes and NGS data. This allowed characterization of non-canonical mutations in BRAF present in adult gliomas; improved estimates of relative frequencies and composition of BRAF alteration types; and integrated clinical-genomic analyses to potentially aid in prognostication.
In this cohort, several important characteristics of BRAF alterations in adult glioma emerge. First, BRAF alterations co-occur with alterations similar to those observed in other cancers, suggesting they may portend similar functional effects in glioma. Second, while overall survival in all patients with Class I alterations was improved relative to other classes, there was no improved survival in GBM. Third, the spectrum of BRAF alterations is different between pediatric and adult glioma, with important ramifications for clinical outcomes. Lastly, BRAF-targeted therapy can benefit a subset of patients with targetable BRAF alterations.
As in other cancers, most Class I BRAF alterations occurred in the relative absence of additional oncogenic alterations, suggesting they are the primary oncogenic drivers in these tumors. We also observed a transcriptional signature of ERK-dependence in tumors with Class I alterations18,23, suggesting potential sensitivity for targeted therapy. Concurrent alterations in other RAS pathway members (NF1, EGFR, etc.) occurred in Class II/III tumors, consistent with the proposed mechanism of ERK activation, while also suggesting ERK-dependence is the primary oncogenic driver in these cancers3,4. Interestingly, gliomas with BRAF gains often exhibited focal chromosome 7q gains, suggesting either an advantageous amplification or that BRAF is a passenger amongst other genes promoting glioma progression24. Of note, 14 tumors in our cohort had hypermutated phenotypes, most commonly with unclassified BRAF alterations. A subset of hypermutated gliomas (23%) had IDH-mutations, which have a known predilection toward hypermutation following temozolomide chemotherapy25. BRAF alterations in hypermutated tumors may be passenger alterations or, as observed recently, may confer a growth advantage26.
Associations between BRAF alteration class and outcome in adults were mixed. Across all patients with Class I alterations, we observed improved overall survival compared to other classes. However, there was no survival advantage among GBM patients with Class I alterations compared to other patients with GBM, suggesting the transition to GBM allows cells to compensate for any growth disadvantage of BRAF-driven ERK dependence27. This phenomenon was also demonstrated in patients with BRAF fusions. While BRAF fusions are associated with an indolent course in pediatric patients, in adults there was a bimodal survival distribution of pilocytic astrocytomas and other higher-grade pathologies with a more aggressive clinical course. This observation underscores the importance of pathologic grade and other clinical features rather than BRAF-alteration alone for prognostication28,29,30,31. Age, CDKN2A/B loss, and molecular markers of GBM were correlated with a worse prognosis, regardless of BRAF alteration, emphasizing their importance for guiding the timing and type of treatment29. Given the retrospective nature of our cohort, most patients did not receive targeted therapy, so it is unclear whether BRAF-targeted therapy has changed the outcomes of patients with GBM11,12. Another nuance to the role of BRAF alterations in glioma prognosis is the broad range of alterations present in adult glioma. Further mechanistic work is necessary to understand whether the BRAF classes observed imply functional pathway dependency and whether they could be exploited with specific therapeutic strategies.
Identification of a targetable BRAF alteration provides patients with novel therapeutic opportunities. In our small retrospective cohort of patients treated with targeted therapy, we observed clinical benefit in a subset of patients. In prospective clinical trials, response rates of 70% and 33% have been observed among adults with low or high grade glioma, respectively11,12. Clinical trials of dimer-disrupting BRAF inhibitors with potential efficacy in additional BRAF-altered classes are ongoing in adults with glioma (NCT05503797). Additionally, improved blood brain barrier penetration may provide additional benefit to patients with central nervous system involvement (NCT04543188). Given the growing list of therapeutic options, accurate identification of BRAF alterations is critical for patient care. Molecular profiling has proven value for accurate tumor diagnoses and is becoming widespread in adults with brain tumors29. In cases where a tissue sample is not feasible, liquid biopsy shows promise to accurately identify targetable alterations such as BRAF from plasma samples32,33.
This landscape of BRAF alterations in adult glioma provides an invaluable resource to clinicians evaluating the functional implications of various BRAF alterations in adults with glioma. Further work is necessary to prospectively validate outcomes and identify the role of other BRAF alterations in patients with glioma. In patients with targetable alterations (Class I–III), small molecule inhibitors can have a significant clinical benefit, underscoring the importance of accurately identifying and classifying BRAF alterations in patients of all ages with glioma.
Methods
Patient data collection
Patients aged ≥18 years at the time of diagnosis with a glioma containing a BRAF alteration on sequencing were identified at the Dana-Farber/Harvard Cancer Center (DFCI), Johns Hopkins Hospital (JHH), and two public repositories: The Cancer Genome Atlas Program (TCGA) and Project Genomics Evidence Neoplasia Information Exchange (GENIE). A single glioma tissue sample collected between 2008–2020 was included per patient (either primary resection or recurrence). For comparison, 90 pediatric (<18 years old) patients with gliomas harboring a BRAF alteration in sequencing data were identified from DFCI, TCGA, and GENIE, with demographic data available for a subset (n = 57). Clinical, histopathologic, and molecular data from tumor tissue were collected and analyzed retrospectively. Medical record review for clinical characteristics and outcomes was performed under protocols approved by the institutional review boards of DFCI and JHH. A waiver of consent was obtained for this retrospective study at Johns Hopkins given infeasibility of consent since many patients were deceased (IRB00243637). Written informed patient consent was obtained for all patients under Dana-Farber Cancer Institute IRB protocol 10–417.
Clinical and pathologic annotation
Pathological diagnosis of tumors within this study was based on the 2021 WHO classification of CNS tumors to the extent possible; however, many cases had insufficient information for precise classification using this system and some broader categories were also used29. Available information on tumor pathologies were reviewed and samples were grouped into seven general pathological categories for further analysis by a neuropathologist (CGE). These categories included glioblastoma, IDH-wildtype (GBM, IDH-WT); astrocytoma, IDH-mutant (Astro, IDH-mt, Grade 2–4); oligodendroglioma (Oligo, IDH-mt, Grade 2–3); pleomorphic xanthoastrocytoma (PXA, Grade 2–3); and pilocytic astrocytoma (PA). A subset of tumors did not fall into one of those diagnostic categories and were grouped based on their grade: other high-grade glioma (HGG; Grade 3–4) and other low-grade glioma (LGG; Grade 1–2). Overall survival (OS) was obtained from public databases or calculated from electronic medical records as time from radiographic diagnosis to date of death. Records from 13 adult patients treated with BRAF-targeted therapy were further reviewed to determine treatment type(s), duration, response, and time to progression(s).
We ran an optimal cut-point analysis using maximally selected rank statistics to identify any age inflection points that significantly corresponded with survival in our overall cohort and found inflection points at 34 and 51 years (Supplementary Fig. 3). Consequently, for the purposes of this study, we defined age interval groups as <18, 18–34, 35–50, and >50 years of age.
Genomic characterization
Genomic mutation profiles were evaluated from panel-based next-generation sequencing assays—which included 227–447 cancer-associated genes (OncoPanel, versions 1–3)34,35 for DFCI samples and 27–435 genes (Neuropath NGS Panel and Solid Tumor Panel versions 2–4)36 for JHH samples—as well as from publicly available targeted mutational profiling microarray sequencing data for 341–398 genes (MSK-IMPACT)31 and exome sequencing data covering 1.06 Mb of cancer-associated genes37 for GENIE and TCGA samples, respectively. Available mutation data were compiled, including single-nucleotide variants (SNV), copy number variants (CNV), and structural rearrangements. In genomic analyses, we incorporated missense mutations, in-frame insertion-deletions (indels), splice site mutations, “truncating” mutations (nonsense mutations, frameshift indels), high-level copy number amplifications38, high-level copy number losses, and structural rearrangements such as gene-gene fusions. Low-copy gains, single-copy deletions, synonymous mutations, and intronic variants were excluded. Genes covered in at least two targeted panels and altered in at least 5% of patients in the combined dataset were included in downstream analysis.
BRAF alterations were classified into six groups based on previously defined functional classes. We used Class I to designate BRAFV600 alterations that activate ERK signaling and are oncogenic as monomers2. Class II and Class III BRAF alterations activate ERK signaling through dimerization that is independent or dependent on upstream RAS activation, respectively3,4. Known pathogenic BRAF rearrangements (“fusions”) and BRAF gene amplifications (“gains”) were also classified into separate categories, with all remaining alterations categorized as “unclassified”.
We parsed hypermutated samples into a distinct category based on total mutational count cutoffs. Tumor mutational burden (TMB) was estimated from targeted sequencing data as the total number of nonsynonymous mutations and indels across the coding regions covered by the gene panels (coverage range between 0.7–1.3 Mb). For downstream analyses, hypermutated samples were defined as those with greater than the 95th percentile TMB (>55 total nonsynonymous mutations).
Statistical analyses
All statistical analyses were performed using R statistical software, version 3.6.3 (R Foundation for Statistical Computing; www.r-project.org). Clustering analysis was performed using 136 genes with cross-coverage across all non-hypermutated samples with genomic data. For each sample, genes were binarized as either altered or non-altered. Logistic principal component analysis (PCA) was implemented using the logisticPCA package (CRAN) to reduce the dimensionality of the binary alterations matrix (genes x samples) and k-means clustering was performed on the first 25 logistic principal components. We used the silhouette and elbow methods to determine the optimal number of k-means clusters.
Correlations between BRAF alteration types, patient characteristics, and other genomic alterations (binarized as present or absent for each gene) were evaluated using Chi-squared test constructed from pairwise contingency tables. Strength of association was assessed using Cramer’s V statistic. Post hoc analysis was applied on significant associations to dissect directions of effect.
Associations between clinical, histopathologic, and molecular covariates of interest and overall survival were assessed by univariate analysis using Cox proportional hazards models. Correction for multiple comparisons was performed using the Benjamini–Hochberg method. Variables with an adjusted false discovery rate (FDR) < 0.05 and altered in at least five individual patients were reported as significant. Overall survival rates were compared by the Kaplan–Meier method with censoring to date of last follow-up and significance determined by log-rank test. All survival analyses were performed using the survival (CRAN) and survminer (CRAN) packages.
RNA sequencing analysis
HTSeq counts matrices39 available for TCGA samples in the cohort were obtained from the public glioblastoma (TCGA-GBM) and low-grade glioma (TCGA-LGG) data repositories. Differential expression analysis was performed using quasi-likelihood F-tests after standard filtering, normalization, and dispersion estimation steps in EdgeR. Pathway analyses were conducted using goseq. Volcano plot and treemap figures were constructed using the EnhancedVolcano and rrvgo R packages. The Benjamini–Hochberg method was used to account for multiple hypothesis testing in both differential expression and pathway analysis.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Datasets from TCGA used in this project are publicly available using the National Cancer Institute Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/). TCGA data are listed under the Project IDs “TCGA-LGG” and “TCGA-GBM.” Datasets from GENIE used in this project are publicly available using the cBioPortal (https://genie.cbioportal.org/) from the “GENIE Cohort v12.0-public” study. Consent for public data sharing was not obtained from DFCI and JHH patients, so datasets from DFCI and Johns Hopkins Hospital are available upon data usage agreement from the corresponding authors.
Code availability
Code used for this analysis is available at https://github.com/pclangat/braf-glioma.
References
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Yaeger, R. & Corcoran, R. B. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 9, 329–341 (2019).
Yao, Z. et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 28, 370–383 (2015).
Yao, Z. et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238 (2017).
Schreck, K. C., Grossman, S. A. & Pratilas, C. A. BRAF mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers 11, 1262 (2019).
Mackay, A. et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e525 (2017).
Ryall, S. et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37, 569–583.e565 (2020).
Hargrave, D. R. et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin. Cancer Res. 25, 7303–7311 (2019).
Perreault, S. et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer 19, 1250 (2019).
Nicolaides, T. et al. Phase I study of vemurafenib in children with recurrent or progressive BRAF(V600E) mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget 11, 1942–1952 (2020).
Kaley, T. et al. BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. J. Clin. Oncol. 36, 3477–3484 (2018).
Wen, P. Y. et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 23, 53–64 (2022).
Vuong, H. G. et al. BRAF mutation is associated with an improved survival in glioma—a systematic review and meta-analysis. Mol. Neurobiol. 55, 3718–3724 (2017).
Da, R., Wang, M., Jiang, H., Wang, T. & Wang, W. BRAF (AMP) frequently co-occurs with IDH1/2, TP53, and ATRX mutations in adult patients with gliomas and is associated with poorer survival than that of patients harboring BRAF (V600E). Front. Oncol. 10, 531968 (2020).
Wang, W., Wang, M., Jiang, H., Wang, T. & Da, R. BRAF(non-V600E) more frequently co-occurs with IDH1/2 mutations in adult patients with gliomas than in patients harboring BRAF(V600E) but without a survival advantage. BMC Neurol. 21, 195 (2021).
McNulty, S. N. et al. BRAF mutations may identify a clinically distinct subset of glioblastoma. Sci. Rep. 11, 19999 (2021).
Pratilas, C. A. et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 68, 9375–9383 (2008).
Dry, J. R. et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 70, 2264–2273 (2010).
Miao, B. et al. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 5, 274–287 (2015).
Cioce, M. & Fazio, V. M. EphA2 and EGFR: friends in life, partners in crime. Can EphA2 be a predictive biomarker of response to anti-EGFR agents? Cancers 13, 700 (2021).
Schreck, K. C., Guajardo, A., Lin, D. D. M., Eberhart, C. G. & Grossman, S. A. Concurrent BRAF/MEK inhibitors in BRAF V600-mutant high-grade primary brain tumors. J. Natl Compr. Cancer Netw. 16, 343–347 (2018).
Lim-Fat, M. J. et al. Clinical, radiological and genomic features and targeted therapy in BRAF V600E mutant adult glioblastoma. J. Neurooncol. 152, 515–522 (2021).
Pratilas, C. A. et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl Acad. Sci. USA 106, 4519–4524 (2009).
Roth, J. J. et al. Whole chromosome 7 gain predicts higher risk of recurrence in pediatric pilocytic astrocytomas independently from KIAA1549-BRAF fusion status. J. Neuropathol. Exp. Neurol. 75, 306–315 (2016).
Yu, Y. et al. Temozolomide-induced hypermutation is associated with distant recurrence and reduced survival after high-grade transformation of low-grade IDH-mutant gliomas. Neuro. Oncol. 23, 1872–1884 (2021).
Campbell, B. B. et al. Mutations in the RAS/MAPK pathway drive replication repair-deficient hypermutated tumors and confer sensitivity to MEK inhibition. Cancer Discov. 11, 1454–1467 (2021).
Johnson, B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
Collins, V. P., Jones, D. T. & Giannini, C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 129, 775–788 (2015).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro. Oncol. 23, 1231–1251 (2021).
Louis, D. N. et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 30, 844–856 (2020).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Kang, K. M. et al. Blood-based detection of BRAF V600E in gliomas and brain tumor metastasis. Cancers 13, 1227 (2021).
Schreck, K. C. S. et al. Preliminary results of binimetinib and encorafenib in adults with recurrent BRAF V600E-mutated hihg-grade glioma. Neuro. Oncol. 24, vii86–vii87 (2022).
Wagle, N. et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2, 82–93 (2012).
Garcia, E. P. et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med. 141, 751–758 (2017).
Palsgrove, D. N. et al. Subependymal giant cell astrocytoma-like astrocytoma: a neoplasm with a distinct phenotype and frequent neurofibromatosis type-1-association. Mod. Pathol. 31, 1787–1800 (2018).
Wang, L. et al. Tumor mutational burden is associated with poor outcomes in diffuse glioma. BMC Cancer 20, 213 (2020).
Beroukhim, R. et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl Acad. Sci. USA 104, 20007–20012 (2007).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Acknowledgements
K.C.S. is supported by the DOD Peer Reviewed Cancer Research Program under Award No. (W81XWH-21-1-0251) and the Johns Hopkins Department of Neurology. Open access publication was supported by the Keep Punching Foundation.
Author information
Authors and Affiliations
Contributions
K.C.S. and W.L.B. conceived of the study and managed the research. C.G.E. confirmed pathology diagnoses and diagnostic categories. P.L., T.L. and E.W. collected and prepared the raw data. P.L., V.M.B., T.L. and E.W. wrote the code. K.C.S., P.L., V.M.B., T.L. and E.W. analyzed the data and created figures. C.A.P. and C.G.E. provided input on study design, manuscript layout, and analyses. K.C.S. and P.L. led the writing of the paper. All authors revised and improved the manuscript and take accountability for the integrity and accuracy of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schreck, K.C., Langat, P., Bhave, V.M. et al. Integrated molecular and clinical analysis of BRAF-mutant glioma in adults. npj Precis. Onc. 7, 23 (2023). https://doi.org/10.1038/s41698-023-00359-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00359-y
