
Abstract
Secondary organic aerosol (SOA) represents a major fraction of atmospheric fine particles. Both biogenic and anthropogenic volatile organic compounds (VOCs) can contribute to SOA through (photo-) oxidation. However, the current understanding of their combined, interactive effect on SOA formation and composition is still limited, challenging the accuracy in assessing global SOA budget, sources, and climate effect. Here we combine laboratory experiments and modelling to show that isoprene can suppress SOA formation from photo-oxidation of anthropogenic aromatics (toluene and p-xylene) with the presence of NOx, and similar SOA suppression phenomena are observed when replacing isoprene with propene. We find that the decreased SOA in such mixed-VOC conditions can be largely attributed to OH scavenging effect, resulting in reduced consumption of parent aromatics. However, various changes in SOA oxidation state (i.e., O/C) and oxidation pathways (i.e., more carbonyls formation) are observed following addition of isoprene, and the SOA chemical composition may not be similar to any single parent hydrocarbon, which implies the existence of complex interactions between the degradation chemistry for alkenes and aromatics. Under the conditions of this work, the OH scavenging effect is largely determined by gas-phase chemistry, which is expected to be widespread in binary or more complex systems in ambient air. More broadly, we infer that the global budget of anthropogenic SOA and its corresponding radiative forcing could be affected by biogenic emission of isoprene, particularly in urban environments with appreciable vegetation coverage.
Similar content being viewed by others


Introduction
The atmospheric (photo-) oxidation of volatile organic compounds (VOCs) emitted from natural and man-made sources leads to the formation of secondary species, notably pollutants like ozone and secondary organic aerosol (SOA)1,2,3. It has been recognised that SOA contributes a significant fraction of ambient tropospheric aerosol4,5, which can cause serious air pollution6, damage human health7, and affect Earth’s climate8,9. The global SOA budget is highly uncertain, and bottom-up estimates of biogenic and anthropogenic SOA fluxes are 12−70 and 2−12 Tg year−1, respectively10, suggesting that global SOA is dominated by biogenic sources.
Under ambient conditions, a variety of hydrocarbons from different sources are SOA precursors and it is expected that photochemical interactions would occur among them during the SOA formation process10,11. However, in current chemical transport models, the parameterisations that describe SOA formation are based on SOA yields from single VOC laboratory experiments, and it is assumed that the yields are additive across VOC mixtures12,13,14,15,16. Compared to single-hydrocarbon laboratory experiments, there have been limited studies to understand how the presence of multiple hydrocarbons from anthropogenic and biogenic sources—each of which is being oxidised to form SOA—alters SOA formation pathways and composition10,16,17,18. Therefore, laboratory studies under mixed-VOC conditions are needed to better understand the sources and formation of ambient SOA, which could help assess the impact of anthropogenic emissions on the environment and climate changes more accurately.
Previous studies19,20,21,22 have shown that biogenic SOA and new particle formation could be suppressed by the presence of isoprene, which may be explained by the competition for OH radicals between isoprene and other compounds. Recently, McFiggans et al.23 found that the suppression of monoterpene SOA yield in the presence of isoprene was caused by the scavenging of both OH and low-volatility products by isoprene and its oxidation products. This finding could be extended more broadly to the real atmosphere that contains a complex mixture of compounds, implying that the global SOA budget from biogenic emissions could be lower than previous estimates24,25. Apart from the interaction between biogenic VOCs, biogenic SOA formation can also be affected by the presence of inorganic anthropogenic species like SO2 and NOx10,11,16,26,27,28. A recent field study in south-eastern USA showed that isoprene-derived SOA production is directly mediated by the abundance of sulphate, while NOx can enhance night-time monoterpene SOA via NO3 oxidation29. All the above studies indicate considerable advances achieved in recent years on understanding of how isoprene and other inorganic anthropogenic pollutants (e.g., SO2, NOx) affect biogenic SOA formation, through laboratory experiments, field measurements and model simulations.
On the other hand, the effect of VOC mixtures on SOA formation is not conclusive and may enhance or suppress SOA in different studies23,30,31,32,33. For example, Li et al.31 found enhanced SOA formation by mixing two anthropogenic VOCs (n-dodecane and 1,3,5-trimethylbenzene) in the presence of NOx and sunlight, and their experiments were conducted in a large-scale outdoor chamber, which is close to the real atmospheric conditions. By contrast, Voliotis et al.32 found that the volatility of SOA from anthropogenic and biogenic VOC mixtures (o-cresol and α-pinene) was in between those measured in single-VOC systems, and their experimental conditions were representative of typical ambient daytime photochemistry with moderate relative humidity (50 ± 5%) and temperature (24 ± 2 °C). Kari et al.33 further conducted chamber experiments by injecting both α-pinene and real-world exhaust from gasoline direct-injection (GDI) engine, and they found that the presence of vehicle emissions may change the oxidation pathways of α-pinene and result in suppression (by more than 50%) of biogenic SOA mass yield. These recent laboratory studies suggested that SOA formation from mixed-VOC condition was governed by the molecular interactions of the oxidation products from different hydrocarbons, which was different and more complex than the SOA derived from single-VOC experiments. However, there are still rather limited laboratory studies on the interactive effect of VOC mixtures from different sources—particularly the effects of biogenic VOC on anthropogenic SOA formation and composition—implying a significant knowledge gap regarding SOA formation chemistry. Given the fact that the real atmosphere is a complex mixture of thousands of organic and inorganic species from various sources1,24,34, an improved understanding of such interaction effects on SOA formation is much needed18,31,35, which consequently motivated the current work.
Aromatics such as toluene and p-xylene are well known as an important class of VOCs with a high SOA yield from anthropogenic emissions such as vehicles and industry36,37, while isoprene is the most abundant biogenic hydrocarbon with global emission of ~500 Tg year−1 38. In this study, we combine chamber experiments and photochemical modelling to investigate the effect of isoprene on anthropogenic SOA formation from toluene and p-xylene. We find a significant suppression of aromatic SOA by isoprene, and further elucidate the underlying chemical mechanisms through additional investigation on analogous reactants, gas-phase and SOA modelling, and SOA composition analysis. Finally, the potential implications on global SOA budget, sources and climate effect from photochemical interaction between anthropogenic and biogenic VOC are discussed.
Results and discussion
Experimental evidence of anthropogenic SOA suppression by isoprene
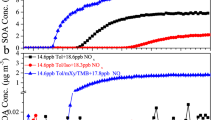
A series of chamber experiments on SOA formation from toluene/NOx and p-xylene/NOx photo-oxidation in the absence and presence of isoprene or propene were performed (see “Methods”), and the initial conditions of each experiment are given in Table 1. The time-dependent SOA mass concentrations and total particle number concentrations are shown in Fig. 1, which is divided into toluene and p-xylene systems. The four toluene/NOx (ZJU095/098/104/130) experiments were regarded as the base conditions, with SOA mass concentrations in the range of 37.5−43.0 μg m−3 and maximum particle numbers of 6.4−9.3 × 104 cm−3 (Fig. 1a, c). Compared with the base conditions, adding similar mixing ratios of isoprene (165 ppbv; ZJU164) or propene (152 ppbv; ZJU176) significantly lowered the particle number, and reduced the aerosol mass loading by up to 80%. The comparison between ZJU164 (165 ppbv isoprene added) and ZJU183 (38 ppbv isoprene added) revealed a lower SOA mass loading and particle number concentration in the former experiment with higher addition of isoprene, indicating that the magnitude of aromatic SOA suppression may depend on isoprene concentrations.

The SOA raw mass concentrations (a, b) and total particle number concentrations (c, d) were derived from SMPS measurements without wall-loss correction. The measured effective densities were applied for converting volume concentration to mass concentration.
Additionally, it was found that the addition of isoprene significantly delayed the onset time of aerosol nucleation, particularly at higher isoprene concentration (e.g., ZJU164) which is in between the two single-hydrocarbon conditions (toluene/NOx and isoprene/NOx). In contrast, the shift of aerosol nucleation onset time was not that obvious for the toluene/propene/NOx experiment (ZJU176). The addition of isoprene resulted in a much lower particle number concentration than when propene was added (ZJU164 vs. ZJU176); since both experiments added similar levels of isoprene (165 ppbv) and propene (152 ppbv), it is suggested that isoprene has a stronger inhibitory effect on new particle formation under such conditions in line with their relative OH reactivity.
As shown in Fig. 1b, compared to the p-xylene/NOx base condition (ZJU220/232), the addition of isoprene (ZJU224) or propene (ZJU230) significantly lowered the aerosol mass concentration by ~50%, consistent with the results of the toluene experiments. As shown in Fig. 1d, the aerosol nucleation onset time was also significantly delayed due to isoprene, whereas the addition of propene resulted in only a very slight delay in aerosol nucleation, which was a similar overall phenomenon to that observed in the toluene experiments. However, both ZJU224 and ZJU230 showed slightly higher maximum particle number concentrations than the base condition, which was not the case for the toluene experiments. This could be related to the weaker aerosol nucleation of p-xylene/NOx than toluene/NOx in these experiments, but the detailed explanation remains unclear.
The SOA mass yields at the end period (7−8 h) of each experiment were calculated according to the wall-loss corrected SOA mass concentrations and reacted hydrocarbons, with the detailed results summarised in Supplementary Table 1. Note that the reacted amount of VOC (ΔVOC) from most of the experiments was obtained from direct VOC measurements (further scaled by initial concentrations), except for four single-hydrocarbon experiments without VOC measurements where simulated VOC decay from SAPRC-18 was used (see more details of gas-phase modelling in the next section). As for mixed-VOC experiments, two yield values (Ymix and Yonly) were calculated based on alternative assumptions to quantify the aerosol mass yield. Ymix was calculated following the assumption that both reacted aromatics and alkenes contributed to SOA mass, while Yonly was calculated by conservatively assuming that only aromatics contributed to SOA mass. For single-hydrocarbon experiments, Yonly alone was calculated because only one parent VOC was in the system.
In the single-hydrocarbon experiments, the average mass yield of toluene SOA was ~22%, while the isoprene SOA yield was 0.78–1.18%; these were both within typical ranges in previous studies12,13,39,40. We found that the addition of isoprene or propene into toluene/NOx could significantly lower the Ymix (by at least ~50% depending on conditions), particularly under high isoprene conditions (ZJU164). This was consistent with the fact that propene is usually not considered to be an SOA precursor and the isoprene SOA yield was also relatively small (0.78–1.18% in this work). Even if it was assumed that toluene was the only contributor to SOA mass in these mixed-VOC experiments, the calculated Yonly was still found to be lower (at least ~15%) than that from toluene-only experiments. Similar results were also found in the p-xylene experiments, where the addition of isoprene or propene clearly lowered the SOA mass yield regardless of whether Ymix or Yonly was calculated.
Modelling gas-phase chemistry and SOA formation
Detailed photochemical modelling was performed to further investigate the reasons behind the observed SOA suppression. Considering that gas-phase chemistry initiates SOA formation, simulations were conducted using five well-established gas-phase chemical mechanisms i.e., SAPRC-07/11/18, CB6 and MCMv3.3.1. As for single-hydrocarbon experiments, Supplementary Figs. 1 and 2 show that almost all the mechanisms were able to explain the measured data to some extent, though some differences among the five mechanisms were observed. This provided a good baseline for mechanistic modelling of more complex conditions. More details about the mechanism performance for the single-hydrocarbon experiments are provided in the Supplementary Discussion.
Here, we focus on the gas-phase results from mixed-VOC experiments (Fig. 2). For toluene/isoprene/NOx experiments, both SAPRC-11 and SAPRC-18 provided reasonable predictions for O3 formation, NO oxidation and hydrocarbon decay, while the other three mechanisms slightly underestimated the O3 formation rate and NO oxidation, particularly under lower isoprene condition (ZJU183). All the mechanisms provided reasonable predictions for the toluene/propene/NOx experiment (ZJU176), though CB6 slightly underestimated the O3 formation. As for the two p-xylene experiments with the addition of isoprene (ZJU224) or propene (ZJU230), the general trends and levels of gas-phase species were correctly captured by SAPRC-11, SAPRC-18 and MCMv3.3.1, though all the mechanisms showed small discrepancies.

Experiments include toluene/isoprene/NOx (ZJU164/183), toluene/propene/NOx (ZJU176), p-xylene/isoprene/NOx (ZJU224) and p-xylene/propene/NOx (ZJU230).
Overall, all the chemical mechanisms were able to predict the gas-phase processing under single or mixed-VOC conditions, particularly the decay process of hydrocarbons under mixed-VOC conditions. Among the five chemical mechanisms, SAPRC-18 modelling results seem to show better agreement with experimental data probably because it considers latest advancement of chemical mechanism development. The simulation results showed that a lower amount of toluene or p-xylene reacted after the addition of isoprene or propene, consistent with the experimental data (Fig. 2 and Supplementary Fig. 2). This is attributed to OH competition by the highly reactive alkene species, consequently resulting in lower OH levels (Supplementary Fig. 3). The lower consumption of toluene or p-xylene due to the addition of isoprene or propene provides a qualitative explanation for the suppression of SOA mass concentration. As indicated by Song et al.41 that the SOA from m-xylene/NOx photo-oxidation is strongly dependent on OH levels in the system, where the addition of propene can decrease OH and slow down the reaction rate of m-xylene. Jaoui et al.42 also found that the addition of isoprene to complex mixture conditions (α-pinene/toluene/NOx) can decrease the amount of reacted toluene and consequently lower the SOA formed.
It has been recognised that the gas-phase autooxidation of peroxy radicals (RO2) can produce highly oxygenated organic molecules (HOMs), which can further contribute to new particle formation and SOA formation43,44. More recently, Heinritzi et al.25 found that the addition of isoprene to the α-pinene oxidation system suppresses nucleation and growth, because the RO2 radicals derived from isoprene and α-pinene are competitive and result in reduced C19–20 dimers and increased C14–15 dimers. Thus, such RO2 termination processes observed in the isoprene and α-pinene mixtures are considered as a critical step that determines the HOMs distribution and subsequent nucleation and growth of SOA. Similar RO2 termination processes are expected to exist in our complex interactions between the degradation chemistry for alkenes and aromatics. Here we further explore the possible connection between SOA formation and RO2 fate under our mixed-VOC conditions. The RO2 removal rates (RO2 + NO and RO2 + HO2 pathways) and concentrations predicted by the MCMv3.3.1 were summarised in Supplementary Fig. 4. For all the experiments in general, the removal pathway of RO2 was overwhelmingly dominated by RO2 + NO in the early period, and gradually shifted to RO2 + HO2 once the NO levels approached ~0. Compared to the base conditions (toluene/NOx or p-xylene/NOx system), the RO2 removal was delayed due to the addition of isoprene, while the addition of propene resulted in an earlier RO2 removal. Such variation of RO2 fate was consistent with the normalised NO decay signal, and may explain the delayed SOA formation due to the addition of isoprene as observed in Fig. 1.
To further simulate SOA formation under mixed-VOC conditions, time-dependent SOA mass yields were calculated for toluene/NOx, isoprene/NOx and p-xylene/NOx experiments. Supplementary Fig. 5 shows the SOA yield growth curves for these three single-hydrocarbon systems, which were specific-fitted using two-product functions12,45. By applying the three specific-fitted yield curves for toluene, isoprene and p-xylene SOA, as well as the reacted VOC amounts (shown in Supplementary Table 1), the predicted SOA mass concentrations for each experiment, including the mixed-VOC experiment, were derived. Figure 3 shows the predicted versus measured SOA mass for the toluene and p-xylene systems. It was found that the SOA mass concentrations predicted by specific-fitted yield curves agreed reasonably well with the measured SOA for almost all single-VOC and mixed-VOC experiments, except for one (p-xylene/isoprene/NOx, ZJU224) that showed some bias outside of the 1:1 line. This suggested that the suppression of SOA mass concentrations observed in the mixed-VOC experiments could be attributed to the decreased consumption of the parent hydrocarbons alone.

The experiments were divided into a toluene and b p-xylene systems. The measured SOA mass concentrations were from SMPS measurements (with wall-loss correction) over 7−8 h. The two-product fitted yield curves (see Supplementary Fig. 5) of specific chamber experiments were derived from their respective toluene/NOx, isoprene/NOx, or p-xylene/NOx experiments; these were used for SOA prediction, particularly for toluene/NOx and p-xylene/NOx with an additional injection of isoprene and propene. Note that VOC measurements were not available for ZJU095/098/104/178, and modelled VOCs from SAPRC-18 were used to derive the reacted amount of VOC (ΔVOC) for these experiments.
As discussed earlier, established gas-phase chemistry can account for the ‘OH scavenging effect’ upon SOA yields induced by isoprene or propene in our mixed-VOC experiments, associated with lower consumption of the parent aromatic hydrocarbons. Gas-phase chemistry and yield-based approaches can provide an overall explanation of SOA formation suppression under the conditions of these experiments, although the chemical systems in our chamber experiments were simplified compared to the complexity of real atmospheric conditions.
SOA chemical composition from AMS measurements
The evolution trends of SOA chemical composition are further investigated in mass spectra (see Supplementary Discussion), Van Krevelen diagram (O/C vs. H/C space) and triangle plots (f43 vs. f44 space; f43 and f44 are AMS organic signal fraction at m/z = 43 and 44, respectively) from AMS measurements. Figure 4a shows the changes in H/C and O/C ratios as a function of OH exposure for each experiment in Van Krevelen diagram. This space has been widely used to interpret the possible involved reactions and addition of functional groups during SOA formation46,47,48, according to the slopes of the straight lines (the slopes of k = 0, −0.5, −1, and −2 are shown in dash lines in Fig. 4a). Specifically, the replacement of an aliphatic carbon (CH2) with a carbonyl group (C(=O)) would result in a slope of −2 in the Van Krevelen diagram, suggesting the ketones or aldehydes formation in the SOA. By contrast, the replacement of a hydrogen with an alcohol group (OH) would only lead to O/C increase, as shown by a slope of 0 in the O/C vs. H/C space. Similarly, the addition of functional group like carboxylic acid (C(=O)OH) during SOA formation would result in a slope of −1, while carboxylic acid formed in the SOA by fragmentation would result in a slope of −0.5 in the evolution of H/C and O/C ratios48. The triangle plots (f43 vs. f44 space) in Fig. 4b can also indicate similar information of SOA evolution processes with less noisy data than those of elemental ratios49,50. Note that the AMS ion signals of m/z 43 and 44 primarily originate from carbonyls and carboxylic acids respectively, with dominant oxygenated fragment ions of C2H3O+ (at m/z = 43) and CO2+ (at m/z = 44)49,51,52.

The SOA data in a Van Krevelen diagram (O/C vs. H/C) and b triangle plot (f43 vs. f44) are coloured by OH exposure. The OH exposure of each experiment was calculated using the average of the predicted OH concentrations from five gas-phase chemical mechanisms (SAPRC-07, SAPRC-11, SAPRC-18, CB6 and MCMv3.3.1). The slopes (k = 0, −0.5, −1, and −2) are shown in dash lines in Van Krevelen diagram47,48. The triangle region in f43 vs. f44 space represents the data for ambient organic aerosol according to Ng et al.49.
As shown in Fig. 4, most of the experiments showed clear evolving trends in both Van Krevelen space and triangle plots as the OH exposure increased. As for toluene/NOx base experiments (ZJU104/130), the H/C and O/C ratios of SOA fall along the slope around −0.5, while f43 remained mostly unchanged as f44 increased in the triangle plots. This suggests that the chemical evolution of toluene SOA (ZJU104/130) is probably dominated by the formation of carboxylic acid, particularly through the fragmentation process. By contrast, the addition of isoprene or propene into toluene/NOx base condition leads to some changes of SOA evolving trends. For example, adding low isoprene of ~38 ppbv (ZJU183) showed both increase of f43 and f44 in the SOA evolution process, suggesting the simultaneous formation of carbonyl compounds and carboxylic acids in this case. ZJU183 further showed a trajectory along the slope of −0.81 in Van Krevelen space, consistent with the above deduction that carbonyl compounds are formed in addition to carboxylic acids. On the other hand, adding high isoprene of ~165 ppbv (ZJU164) did not show a clear evolving trajectory, with the H/C and O/C ratios distributing in a narrow range. This case is similar to the pattern of isoprene/NOx (ZJU189), as both experiments have similar level of initial isoprene and relatively low SOA loadings. The addition of ~152 ppbv propene (ZJU176) showed a trajectory of H/C and O/C ratios along the slope around −1, while f43 remained mostly unchanged as f44 increased in the triangle plots. This suggests that the formation route of carboxylic acid in ZJU176 may shift to some extent from fragmentation to functionalization due to the addition of propene. Besides, both f44 and O/C represent the oxidation state of SOA. The addition of isoprene or propene both reduced the magnitude of the increase in f44 and O/C during an experiment. Table 1 further summarises the typical chemical parameters of SOA at the end of each experiment. A decrease in f44 and O/C, as well as an increase in H/C were observed due to the addition of isoprene or propene, which agreed with the results shown in Van Krevelen space in Fig. 4a. Such phenomena indicated that the oxidation state of SOA composition was reduced under mixed-VOC conditions, probably because of (1) reduced OH exposure due to the OH scavenging effect and (2) the aforementioned changes in the oxidation pathways and chemical composition of SOA.
Compared to the p-xylene/NOx base condition (ZJU220/232), the addition of isoprene (ZJU224) or propene (ZJU230) leads to a more discrete pattern in the evolving trend in the H/C vs. O/C space in Fig. 4a. This phenomenon is similar to the narrow distributing of H/C and O/C ratios in the toluene/isoprene/NOx (ZJU164) and toluene/propene/NOx (ZJU176) experiments. Unlike the toluene/NOx base experiments, p-xylene/NOx base experiments (ZJU220/232) showed increase of both f43 and f44 in the SOA evolution process, suggesting the simultaneous formation of carbonyl compounds and carboxylic acids in p-xylene case. As observed in the f43 vs. f44 space in Fig. 4b, the addition of isoprene (ZJU224) resulted in less increase of f44 as f43 increased, indicating relatively more carbonyl compounds formation due to the addition of isoprene. This is consistent with the results from toluene/isoprene/NOx experiment of low isoprene concentration (ZJU183) in the earlier discussion, where more carbonyl formation was evidenced by the changed slope in Van Krevelen space and increased f43 trends in triangle plots. The addition of propene (ZJU230) showed a changed trajectory in the low OH exposure in triangle plots, and reason for this phenomenon is still not clear and requires further investigation.
Overall, we find that the addition of isoprene or propene could not only suppress the aromatic SOA mass and yield, but also change the oxidation state and chemical composition of SOA. The addition of isoprene into aromatic/NOx photo-oxidation may reduce the magnitude of oxidation state increase during the experiment and result in SOA formation with more carbonyl compounds, though these are qualitative and the extent of such changes could depend on the addition amount of isoprene. The current AMS analysis also indicates the complexity of SOA composition under such mixed-VOC conditions, which implies that a detailed molecular characterisation of SOA composition is required in future studies.
Atmospheric implications
Aromatics are well known as an important class of anthropogenic VOCs from vehicle and industrial emissions, and many previous chamber studies12,13 have confirmed their high SOA yields in the presence of NOx and sunlight. Therefore, it is expected that aromatic hydrocarbons can contribute to urban fine particles through photochemical processes. The present study shows that the aromatic SOA mass yields were suppressed by the addition of isoprene or propene, which could be attributed to the OH scavenging effect that lowers consumption of the parent aromatics. Considering the reasonably good performance of the five different chemical mechanisms (SAPRC-07/11/18, CB6, and MCMv3.3.1) in simulating our experiments, we further demonstrated that the suppression of aromatic SOA could be quantitatively explained by the current gas-phase chemistry and yield-based SOA predictions. Such OH scavenging effect proposed by McFiggans et al.23 could be widespread in binary or more complex mixture conditions. Our current experiments are performed under high-NOx conditions that are representative of the daytime photochemistry, and the OH production in such reaction system is limited due to the radical recycles (e.g., HO2 + NO → OH + NO2). However, it still requires more investigations to examine whether the OH scavenging effect holds in reaction systems with surplus OH (e.g., low-NOx conditions), where adding isoprene may have limited influences on the OH consumption. Note that the product scavenging effect (removal of oxidation products with high SOA-formation potential) further proposed by McFiggans et al.23 might also exist in our mixed-VOC conditions; this could further prevent the multi-generation oxidation process, as evidenced by the decreased O/C of SOA due to the addition of isoprene in our toluene experiments. Unfortunately, due to limitations over measurements of oxidised products, this cannot be examined in greater depth in the present study.
The findings of this study imply that the global budget of anthropogenic SOA might be significantly affected by biogenic emissions such as isoprene, particularly in some urban environments with large coverings of land vegetation where both anthropogenic and biogenic emissions are mixed. More broadly, this study further revealed the critical importance of the interaction between aromatics and short-chained alkenes on SOA formation: combining aromatics (higher SOA yield and lower OH rate constant (kOH)) and alkenes (lower SOA yield and higher kOH) in the presence of NOx and sunlight could result in a lower SOA mass. Accounting for such interactive effects can provide new insights for improving the SOA module of chemical transport models, which will allow us to achieve an improved understanding of SOA sources under real-world conditions. Additionally, anthropogenic SOA is well-recognised as an important component for evaluating the impact of human activities on climate change. If the suppression by isoprene on the global budget of anthropogenic SOA is considered and parameterised in current climate models, the corresponding magnitude of the impact of human activities on radiative forcing might also change. Given that both anthropogenic and biogenic VOC emissions will continue to change over time, it is also expected that the aerosol-climate interaction will present a highly dynamic system in the future; this highlights the importance of better understanding SOA formation derived from VOC mixtures of different sources.
The current AMS analysis also showed some interesting and highly variable phenomena related to the chemical characteristics of SOA, particularly the changed oxidation state (i.e., O/C) and oxidation pathways (i.e., more carbonyls formation) under mixed-VOC conditions, which are still not fully understood at present. This implies that SOA formation under mixed-VOC conditions likely undergoes highly complex chemical processes between the degradation chemistry for the alkenes and aromatics. However, parameterisation of such interactive effect in models requires additional work, and it relies on the ability of modelling the transformation from gas-phase species to SOA with fundamental understanding and representation rather than adjustments to fit chamber data. In addition to the daytime photochemistry, nighttime NO3 oxidation can also be a dominant pathway of SOA under certain circumstances, and SOA formation from VOC mixtures through NO3 oxidation is relevant to the real atmosphere but not yet understood. We suggest that more comprehensive mixed-VOC experiments with high quality data and molecular characterisation of SOA composition should be conducted to further explore the interaction between anthropogenic and biogenic VOC mixtures and their combined effect on SOA formation, which should cover a wide range of experimental conditions that are more representative of real-world conditions.
Methods
Experiment summary
The CAPS-ZJU (Complex Air Pollution Study-Zhejiang University) smog chamber is a 3 m3 Teflon-FEP chamber housed in a well-controlled enclosure, with 60 controlled ultraviolet (UV) lamps providing the light source53,54. A total of 15 smog chamber experiments across a broad range of scenarios were performed to support the objectives of this study, with the initial concentrations of VOCs and NOx in each experiment given in Table 1. Specifically, toluene/NOx and p-xylene/NOx experiments were used as two base conditions, as toluene and p-xylene are well recognised as typical precursors of anthropogenic SOA. Isoprene was the major focus of this study, and three experiments (ZJU164/183/224) were conducted with different levels of isoprene added into the above base conditions; this aimed to address the main issue of how isoprene affects anthropogenic SOA formation. In addition, since propene is a typical reactive hydrocarbon and usually not regarded as an SOA precursor, two experiments (ZJU176/230) were performed by adding propene into the base conditions instead of isoprene, assisting explain the isoprene addition case considering some chemical similarities between the two short-chained reactive alkenes. The abbreviations ‘tol’, ‘pxyl’, ‘isop’, and ‘prop’ for toluene, p-xylene, isoprene, and propene, respectively, are used elsewhere. Note that some parallel single-hydrocarbon experiments were conducted under similar conditions to confirm the reproducibility of the results.
Each experiment was performed in a 7−8 h photochemical window at equivalent light intensity (JNO2) of 0.36−0.40 min−1. All the experiments were conducted under dry conditions (relative humidity (RH) < 10%) and in the absence of seed aerosol. We choose low RH ( < 10%) in our chamber experiments to avoid and exclude the complex influence (e.g., multiphase chemistry) under high humidity condition. Upon turning on the UV lights, the temperature inside the chamber increased from ~20 to ~35 °C within 2 h due to the heat released from the lights, and then stabilised at ~35 °C until the end of the experiment. All experiments had similar temperature profiles, which ensured that the experiments were comparable and repeatable54. The background air was supplied by a Zero Air generator (Aadco 737-15, USA). After each experiment, the chamber was continually flushed with clean air for at least 12 h to ensure the particle number concentration below 10 cm−3 and the reactive gas concentration below 1 ppbv. More details of the chamber characterisation and experimental methods can be found in our recent publication54.
Instrumentation and analysis
The CAPS-ZJU chamber facility was equipped with a suite of state-of-the-art instruments to characterise the chemical composition, mass concentration, size distribution, effective density, gas concentration, and other physicochemical properties of particles. Specifically, the NOx and O3 concentrations were measured using commercial gas analysers (Thermo Scientific 42i and 49i, USA). The gaseous hydrocarbons were measured by a gas chromatography-mass spectrometer (GC–MS; Agilent 7890B–5977 A, USA) coupled with an automated pre-concentrator (Entech 7100A, USA). The particle size distribution (14–700 nm) was determined by a scanning mobility particle sizer (SMPS, TSI 3936) with a long differential mobility analyser (DMA, TSI 3081) in combination with a butanol condensation particle counter (CPC, TSI 3776). An aerosol particle mass analyser (APM, Kanomax 3601) was used to measure particle effective density by coupling the DMA, APM, and CPC. As summarised in Supplementary Table 2, the effective density of SOA measured by DMA-APM-CPC ranged from 1.27 to 1.36 g cm−3 depending on the reaction system, which was also close to the density range of typical SOA reported in previous literature55,56. Then, the aerosol mass concentration was derived from the particle number size distribution. Size-dependent aerosol wall-loss correction was performed to account for the particles deposited onto the walls, following the method applied in our previous work54.
The composition of SOA was determined by a high-resolution time of flight aerosol mass spectrometry (HR-ToF-AMS, hereafter AMS; Aerodyne Research Inc., USA), with method described in detail in our previous studies57,58. In brief, the AMS was operated in the high sensitivity V-mode and the high resolution W-mode every 2 min, alternately. The AMS data was analysed by SQUIRREL v1.57l and PIKA v1.16l software written into Igor Pro 6.3.7.2 (WaveMetrics Inc., USA). The elemental ratios (i.e., H/C and O/C) of SOA were determined using the Improved-Ambient (IA) method developed by Canagaratna et al.59. More details of AMS operation and analysis were described in the Supplementary Method.
Gas-phase chemistry and SOA modelling
The gas-phase chemistry box models used for this study are outlined in Supplementary Table 3. SAPRC and CB (carbon bond) mechanisms are widely used as core components for chemical transport models. The specific versions used in this study were SAPRC-07, SAPRC-11, SAPRC-18 and CB6, and have been applied in our recent work60. SAPRC-18 is the latest version of SAPRC mechanism (release in May 2020, see https://intra.cert.ucr.edu/~carter/SAPRC/18/), which has a significant update with alkene chemistry and considers the H-shift isomerizations of peroxy radicals. Note that all the SAPRC series mechanisms (SAPRC-07/11/18) used in this study were detailed versions with explicit representation of individual VOC degradation chemistry61,62,63. There are simplified versions of SAPRC by lumping the VOC species for chemical transport modelling, while the detailed versions of SAPRC are widely used for mechanism evaluation utilising environmental chamber data; more details are available at http://www.cert.ucr.edu/~carter/SAPRC/. CB6 was developed as a lumped mechanism for simplicity64, with the least chemical details among the five mechanisms. The Master Chemical Mechanism (MCMv3.3.1) is a near-explicit mechanism with more detailed reactions (http://mcm.leeds.ac.uk/MCM/)65,66. The smog chamber experiments were simulated using the above five mechanisms with the additional CAPS-ZJU chamber-specific auxiliary wall-mechanism, which has been characterised in detail in our recent publication54.
The SOA mass yield is defined as the mass of SOA produced per unit of hydrocarbon reacted, which has been widely used to predict SOA formation12,45. In this study, yield curves were obtained from the single-hydrocarbon experiments by fitting the time-dependent SOA mass yield to aerosol mass concentration, which could be used to predict SOA formed by toluene, isoprene, and p-xylene, respectively. Combined with the measured VOC decay, these specific-fitted yield curves were further used to predict SOA formation under mixed-VOC conditions.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author (likangweizju@foxmail.com) on reasonable request.
References
Atkinson, R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 34, 2063–2101 (2000).
Ziemann, P. J. & Atkinson, R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem. Soc. Rev. 41, 6582–6605 (2012).
Edwards, P. M. et al. Transition from high- to low-NOx control of night-time oxidation in the southeastern US. Nat. Geosci. 10, 490–495 (2017).
Zhang, Q. et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 34, L13801 (2007).
Jimenez, J. L. et al. Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 (2009).
Huang, R. et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 514, 218–222 (2014).
Shiraiwa, M. et al. Aerosol health effects from molecular to global scales. Environ. Sci. Technol. 51, 13545–13567 (2017).
Seinfeld, J. H. et al. Improving our fundamental understanding of the role of aerosol-cloud interactions in the climate system. Proc. Natl Acad. Sci. USA 113, 5781–5790 (2016).
Shrivastava, M. et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophys. 55, 509–559 (2017).
Hallquist, M. et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 9, 5155–5236 (2009).
Kroll, J. H. & Seinfeld, J. H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 42, 3593–3624 (2008).
Ng, N. L. et al. Secondary organic aerosol formation from m-xylene, toluene, and benzene. Atmos. Chem. Phys. 7, 3909–3922 (2007).
Hildebrandt, L. et al. High formation of secondary organic aerosol from the photo-oxidation of toluene. Atmos. Chem. Phys. 9, 2973–2986 (2009).
Lane, T. E. et al. Simulating secondary organic aerosol formation using the volatility basis-set approach in a chemical transport model. Atmos. Environ. 42, 7439–7451 (2008).
Henze, D. K. et al. Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: High- vs. low-yield pathways. Atmos. Chem. Phys. 8, 2405–2420 (2008).
Carlton, A. G. et al. A review of Secondary Organic Aerosol (SOA) formation from isoprene. Atmos. Chem. Phys. 9, 4987–5005 (2009).
Hildebrandt, L. et al. Evaluating the mixing of organic aerosol components using high-resolution aerosol mass spectrometry. Environ. Sci. Technol. 45, 6329–6335 (2011).
Emanuelsson, E. U. et al. Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties. Atmos. Chem. Phys. 13, 2837–2855 (2013).
Kiendler-Scharr, A. et al. New particle formation in forests inhibited by isoprene emissions. Nature 461, 381–384 (2009).
Lee, S. et al. Isoprene suppression of new particle formation: Potential mechanisms and implications. J. Geophys. Res. Atmos. 121, 14614–635 (2016).
Kanawade, V. P. et al. Isoprene suppression of new particle formation in a mixed deciduous forest. Atmos. Chem. Phys. 11, 6013–6027 (2011).
Kiendler-Scharr, A. et al. Isoprene in poplar emissions: Effects on new particle formation and OH concentrations. Atmos. Chem. Phys. 12, 1021–1030 (2012).
McFiggans, G. et al. Secondary organic aerosol reduced by mixture of atmospheric vapours. Nature 565, 587–593 (2019).
Yu, F. Atmospheric reaction networks affecting climate are more complex than was thought. Nature 565, 574–575 (2019).
Heinritzi, M. et al. Molecular understanding of the suppression of new-particle formation by isoprene. Atmos. Chem. Phys. 20, 11809–11821 (2020).
Wennberg, P. O. et al. Gas-phase reactions of isoprene and its major oxidation products. Chem. Rev. 118, 3337–3390 (2018).
Surratt, J. D. et al. Evidence for organosulfates in secondary organic aerosol. Environ. Sci. Technol. 41, 517–527 (2007).
Shrivastava, M. et al. Urban pollution greatly enhances formation of natural aerosols over the Amazon rainforest. Nat. Commun. 10, 1046 (2019).
Xu, L. et al. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. Proc. Natl Acad. Sci. USA 112, 37–42 (2015).
Ahlberg, E. et al. Secondary organic aerosol from VOC mixtures in an oxidation flow reactor. Atmos. Environ. 161, 210–220 (2017).
Li, J. et al. Enhanced secondary organic aerosol formation from the photo-oxidation of mixed anthropogenic volatile organic compounds. Atmos. Chem. Phys. 21, 7773–7789 (2021).
Voliotis, A. et al. Exploring the composition and volatility of secondary organic aerosols in mixed anthropogenic and biogenic precursor systems. Atmos. Chem. Phys. 21, 14251–14273 (2021).
Kari, E. et al. Potential dual effect of anthropogenic emissions on the formation of biogenic secondary organic aerosol (BSOA). Atmos. Chem. Phys. 19, 15651–15671 (2019).
Seinfeld, J. H. & Pandis, S. N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change (John Wiley & Sons, 2016).
Hoyle, C. R. et al. A review of the anthropogenic influence on biogenic secondary organic aerosol. Atmos. Chem. Phys. 11, 321–343 (2011).
Derwent, R. G. et al. Secondary organic aerosol formation from a large number of reactive man-made organic compounds. Sci. Total Environ. 408, 3374–3381 (2010).
Peng, J. et al. Gasoline aromatics: A critical determinant of urban secondary organic aerosol formation. Atmos. Chem. Phys. 17, 10743–10752 (2017).
Guenther, A. B. et al. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions. Geosci. Model Dev. 5, 1471–1492 (2012).
Dommen, J. et al. Laboratory observation of oligomers in the aerosol from isoprene/NOx photooxidation. Geophys. Res. Lett. 33, 338–345 (2006).
Kroll, J. H. et al. Secondary organic aerosol formation from isoprene photooxidation under high-NOx conditions. Geophys. Res. Lett. 32, L18808 (2005).
Song, C. et al. Impact of propene on secondary organic aerosol formation from m-xylene. Environ. Sci. Technol. 41, 6990–6995 (2007).
Jaoui, M. et al. Formation of secondary organic aerosol from irradiated α-pinene/toluene/NOx mixtures and the effect of isoprene and sulfur dioxide. J. Geophys. Res. Atmos. 113, D09303 (2008).
Schervish, M. & Donahue, N. M. Peroxy radical chemistry and the volatility basis set. Atmos. Chem. Phys. 20, 1183–1199 (2020).
Bianchi, F. et al. Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: A key contributor to atmospheric aerosol. Chem. Rev. 119, 3472–3509 (2019).
Odum, J. R. et al. Gas/particle partitioning and secondary organic aerosol yields. Environ. Sci. Technol. 30, 2580–2585 (1996).
Chen, Q. et al. Elemental composition of organic aerosol: The gap between ambient and laboratory measurements. Geophys. Res. Lett. 42, 4182–4189 (2015).
Heald, C. L. et al. A simplified description of the evolution of organic aerosol composition in the atmosphere. Geophys. Res. Lett. 37, L08803 (2010).
Sato, K. et al. AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1,3,5-trimethylbenzene in the presence of NOx: Effects of chemical structure on SOA aging. Atmos. Chem. Phys. 12, 4667–4682 (2012).
Ng, N. L. et al. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 10, 4625–4641 (2010).
Zhao, D. et al. Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene. Atmos. Chem. Phys. 18, 1611–1628 (2018).
Ng, N. L. et al. Changes in organic aerosol composition with aging inferred from aerosol mass spectra. Atmos. Chem. Phys. 11, 6465–6474 (2011).
Liu, T. et al. Comparison of secondary organic aerosol formation from toluene on initially wet and dry ammonium sulfate particles at moderate relative humidity. Atmos. Chem. Phys. 18, 5677–5689 (2018).
Li, K. et al. Smog chamber study on aging of combustion soot in isoprene/SO2/NOx system: Changes of mass, size, effective density, morphology and mixing state. Atmos. Res. 184, 139–148 (2017).
Li, K. et al. Characterization of a new smog chamber for evaluating SAPRC gas-phase chemical mechanism. J. Environ. Sci. -China 95, 14–22 (2020).
Sato, K. et al. Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons. Atmos. Environ. 44, 1080–1087 (2010).
Nakao, S. et al. Density and elemental ratios of secondary organic aerosol: Application of a density prediction method. Atmos. Environ. 68, 273–277 (2013).
Chen, L. et al. The effects of humidity and ammonia on the chemical composition of secondary aerosols from toluene/NOx photo-oxidation. Sci. Total Environ. 728, 138671 (2020).
Li, K. et al. Chemical characteristics and sources of PM1 during the 2016 summer in Hangzhou. Environ. Pollut. 232, 42–54 (2018).
Canagaratna, M. R. et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications. Atmos. Chem. Phys. 15, 253–272 (2015).
Li, K. et al. Evaluation of a new chemical mechanism for 2-amino-2-methyl-1-propanol in a reactive environment from CSIRO smog chamber experiments. Environ. Sci. Technol. 54, 9844–9853 (2020).
Carter, W. P. Development of the SAPRC-07 chemical mechanism. Atmos. Environ. 44, 5324–5335 (2010).
Carter, W. P. & Heo, G. Development of revised SAPRC aromatics mechanisms. Atmos. Environ. 77, 404–414 (2013).
Carter, W. P. Documentation of the SAPRC-18 Mechanism. Report to California Air Resources Board Contract No. 11-761. https://intra.engr.ucr.edu/~carter/SAPRC/18/S18doc (2020).
Yarwood, G. et al. Development, Evaluation and Testing of Version 6 of the Carbon Bond Chemical Mechanism (CB6). Final Report to Texas Commission on Environmental Quality. https://www.tceq.texas.gov/assets/public/implementation/air/am/contracts/reports/pm/5820784005FY1026-20100922-environ-cb6.pdf (2010).
Bloss, C. et al. Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons. Atmos. Chem. Phys. 5, 641–664 (2005).
Jenkin, M. E. et al. The MCM v3.3.1 degradation scheme for isoprene. Atmos. Chem. Phys. 15, 11433–11459 (2015).
Acknowledgements
We thank Zhier Bao, Lixia Han, Xingya Zhao, and Yanyun Zhao for their assistance with the experiments. We also thank Chunshan Liu of Beijing Convenient Environmental Tech Co. Ltd for his help and support in smog chamber setup. This work was supported by Ministry of Science and Technology PR China (No. G2021060002L).
Author information
Authors and Affiliations
Contributions
K.L. conceived the study; K.L., X.Z, and C.L. performed experiments; K.L. performed the modelling; K.L., X.Z., and B.Z. analysed data; K.L., B.Z., and Z.B. wrote the paper; W.J.B., S.W., H.Y., L.C., C.Geng, W.Y., A.M., and C.George helped to interpret the results and revised the manuscript. All authors contributed to the manuscript preparation and discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K., Zhang, X., Zhao, B. et al. Suppression of anthropogenic secondary organic aerosol formation by isoprene. npj Clim Atmos Sci 5, 12 (2022). https://doi.org/10.1038/s41612-022-00233-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41612-022-00233-x
This article is cited by
-
Secondary organic aerosol formation from mixed volatile organic compounds: Effect of RO2 chemistry and precursor concentration
npj Climate and Atmospheric Science (2022)
