
Abstract
Colistin remains one of the last-resort therapies for combating infections caused by multidrug-resistant (MDR) Enterobacterales, despite its adverse nephro- and neuro-toxic effects. This study elucidates the mechanism of action of a non-antibiotic 4-anilinoquinazoline-based compound that synergistically enhances the effectiveness of colistin against Salmonella enterica. The quinazoline sensitizes Salmonella by deactivating intrinsic, mutational, and transferable resistance mechanisms that enable Salmonella to counteract the antibiotic impact colistin, together with an induced disruption to the electrochemical balance of the bacterial membrane. The attenuation of colistin resistance via the combined treatment approach also proves efficacious against E. coli, Klebsiella, and Acinetobacter strains. The dual therapy reduces the mortality of Galleria mellonella larvae undergoing a systemic Salmonella infection when compared to individual drug treatments. Overall, our findings unveil the potential of the quinazoline-colistin combined therapy as an innovative strategy against MDR bacteria.
Similar content being viewed by others
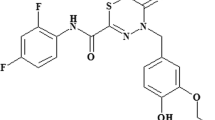


Introduction
Antibiotic resistance is one of the most important threats to human health worldwide and it is concomitant with the urgency of developing new effective antimicrobial compounds. The acquisition of antibiotic resistance mechanisms by bacteria has been attributed to the selective pressure imposed by environmental, human and animal healthcare, general overuse and misuse of these compounds, and to the scarcity in drug development by the pharmaceutical industry1,2.
Bacterial infections are commonly treated with bacteriostatic or bactericidal drugs. Although very effective, alternatives, such as the development of anti-virulence agents that block key pathogenesis mechanisms (pathoblockers), are promising strategies to diminish the emergence of antibiotic resistance mechanisms3. In addition, alternative new drugs can act as adjuvants and reduce the effective concentration of commonly used antibiotics and minimize the emergence of resistance.
In this context, the WHO has classified Salmonella as a high-priority group for the development of new antibacterial therapies4,5. Salmonella senses environmental changes and responds to the detection of host cues by triggering a virulence program. PhoP/PhoQ is a signal transduction cascade that belongs to the bacterial family of two-component regulatory systems (TCS) and has an essential role during the infection of Salmonella into the mammalian host. This TCS consists of PhoQ, a transmembrane sensor with bifunctional histidine kinase/phosphatase activity, and PhoP, a cytoplasmic response regulator6. The system responds to input signals that bacteria encounter during their life cycle, such as availability of Mg2+, presence of long chain unsaturated fatty acids or cationic antimicrobial peptides (CAMPs) and acidic pH7,8,9 As adaptive output responses, PhoP/PhoQ controls the expression of genes involved in magnesium homeostasis10,11, LPS-modifications12, resistance to acidic pH13 the internalization and survival within either phagocytic or non-phagocytic cells14.
The PhoP/PhoQ system has a crucial role during the life cycle of Salmonella and the absence of TCS in mammals, make this TCS an optimal target to develop new antimicrobial therapies15. In this regard, we have recently identified two quinazoline-based compounds which negatively regulate PhoP-activated genes by targeting the PhoQ histidine-kinase activity16.
Colistin is a member of the polymyxins that are cationic antimicrobial oligopeptides composed of a cyclic heptapeptide linked to a tripeptide acylated at its N-terminus by a fatty acid17. These antibiotics disorganize the bacterial envelope in two stages: (1) they interact electrostatically with the negative charges of lipid A, displacing Ca2+ and Mg2+ that stabilize LPS, and (2) the acylated chain inserts within the outer membrane (OM) with formation of pores, promoting the own entry of the molecule into the periplasm. Then, the antibiotic accesses the inner membrane (IM) by including hydrophilic groups in the fatty acid chains18. In addition, Sabnis et al19 demonstrated that modified LPS precursors that would be transported from the IM to the OM are also target of colistin, leading to cell lysis17. Polymyxin E, known as colistin, is a potent clinical antibiotic whose use nearly ceased in the early 1990s due to nephrotoxicity and neurotoxicity. In recent years, it has returned as a last resort antibiotic, due to its effectiveness against multi-drug resistant Gram-negative pathogens17. The veterinary use of colistin allows for the selection of of colistin resistance acquisition in zoonotic bacteria strains, such as non-typhoidal serovars of Salmonella enterica, and this mediates the spread of resistance mechanisms along the food chain20.
PhoP/PhoQ TCS activates the expression of genes that introduce modifications in LPS. This allows bacteria to counteract the bactericidal effect of polymyxins. In the presence of sub-inhibitory concentrations of colistin, the system promotes the expression of pmrD, whose gene product activates the PmrA/PmrB TCS and induces polymyxin resistance genes (ugd, pmrCAB, pgbPE)12.
Lipid A is the LPS target of the PhoP/PhoQ-dependent modifications: (1) introduction of 4-amino-4-deoxy-L-arabinose (L-Ara4N) in the phosphate at position 4, controlled by the pmr, arn or pbg operons12 and (2) introduction of phosphoethanolamine (PEtN) in the position 1-phosphate, controlled by pmrC21,22 Ugd, an UDP-glucose dehydrogenase under PhoP, PmrA and Rcs-dependent regulation, plays a crucial role in L-Ara4N pathway since it converts UDP-glucose to UDP-glucuronic acid, the substrate for the arn pathway23,24.
In this study, we demonstrate how the quinazoline CA439 attenuates the expression of genes that define colistin resistance and thus, it increases colistin efficacy. The quinazoline-mediated enhancement of the polymyxin antibiotic capacity does not only rely on the PhoP/PhoQ-dependent resistance mechanisms inactivation but also on the concomitant alteration of electrochemical properties of the bacterial membrane. The dual treatment attenuates Salmonella virulence in an in vivo model of infection. In sum, our results position the combined colistin-quinazoline use as a promising option to develop a therapeutic strategy to fight against Salmonella infections.
Results
CA439 potentiates the bactericidal effect of colistin
Salmonella typhimurium ATCC 14028 (STM14028) was used as a study model throughout this work. 4-anilinoquinazoline-based compounds down-regulate the activity of the S. typhimurium PhoP/PhoQ system by acting as competitive inhibitors of ATP in the PhoQ autophosphorylation reaction16. We selected CA439 (GI262866A) as representative of these quinazolines (synthesized as previously described16) to evaluate its ability to potentiate colistin activity. All assays were performed in LB, a low Mg2+ medium that ensures the PhoP/PhoQ system with an inducing environmental condition7,8,9,10. CA439 effect was evaluated up to 25 μM, a sub-inhibitory concentration for Salmonella growth capacity that showed a repressive effect over PhoP/PhoQ activity16. The checkerboard assay was employed in which the fractional inhibitory concentration (FIC) was calculated25. The dose-dependent enhancing effect of colistin by CA439 reduced the MIC of colistin from 2.5 μg mL−1 to 0.312 µg mL−1, and resulted in a FIC of 0.125 (Fig. 1a, left). This was corroborated by counting colony-forming units (Fig. 1a, right). None of the CA439-analogues (CA176, CA209, CA454 and CA490, previously synthesized and characterized16) that exerted no repressive effect over PhoP/PhoQ activity showed a colistin-enhancing effect (Fig. 1b), reinforcing that the effect of CA439 selectively targets PhoQ histidine kinase.

Susceptibility to colistin can be increased by combination with CA439. Checkerboard assay showing the combination of colistin with (a) increasing doses of CA439 or (b) 25 μM of CA-analogues that do not inhibit the PhoP/PhoQ system. Dark blue regions represent higher cell density. Data represent the mean OD (600 nm) of at least three biological replicates. Right panel shows the CFU counting obtained in each growth condition: (a) DMSO (LB-medium with the addition of DMSO), CA439 (25 μM CA439 alone), colistin (0.312 μg mL−1 colistin alone) and CA439 + colistin (25 μM CA439 + 0.312 μg mL−1 colistin) and (b) 25 μM of each compound with or without 1.25 μg mL−1 colistin. Fractional inhibitory concentration (FIC) was obtained by dividing the colistin MIC in the presence of CA439 (0.312 μg mL−1) by the colistin MIC in the absence of CA439 (2.5 μg mL−1). Statistical analysis was performed (a) using one-way ANOVA with Tukey’s correction multiple comparison test (****P < 0.0001; ns, no significant differences) and (b) unpaired t test for each compound assayed (ns, no significant difference).
CA439 is able to access the bacterial cytoplasm as it interacts with the catalytic domain of PhoQ16. Despite this, we hypothesized if a permeabilizing effect of sublethal concentrations of colistin could allow to build-up an intracellular concentration of quinazoline that was lethal to Salmonella. No potentiating effect was observed when CA439 and either EDTA or SDS (OM-permeabilizing agents that do not promote LPS modifications) were combined (Supplementary Fig. S1), suggesting that CA439-colistin potentiation does not rely on the colistin-mediated entry of the quinazoline by an OM permeabilizing action.
CA439-colistin enhancing effect is partially PhoP/PhoQ-dependent
To assess whether the observed phenotype depends exclusively on the inhibition of the PhoP/PhoQ system by CA439, single mutants in the response regulator (ΔphoP) or in the histidine kinase (ΔphoQ), and a ΔphoPQ mutant strain were tested (Fig. 2). The colistin-CA439 enhancing phenotype was reduced by twofold in these strains (a FIC of 0.25 for mutant strains versus a FIC of 0.125 for the wild type strain), indicating that CA439 exerts an additional action alongside with the effect on PhoP/PhoQ. Both mutant strains were tested in the presence of CA439 inactive analogues, and no potentiation effect was observed (Supplementary Fig. S2), indicating that the CA439 enhancing effect is specific to this compound.

CA439 enhancement of the bactericidal effect of colistin is partially PhoP/PhoQ-dependent. Checkerboard assay showing CA439-colistin combination in (a) phoP mutant, (b) phoQ mutant and (c) phoPQ mutant. Dark blue regions represent higher cell density. Data represent the mean OD (600 nm) of at least three biological replicates. Right panel shows the CFU counting obtained in each growth condition: DMSO (LB-medium with the addition of DMSO), CA439 (25 μM CA439 alone), colistin (0.312 μg mL−1 colistin alone) and CA439 + colistin (25 μM CA439 + 0.312 μg mL−1 colistin). Fractional inhibitory concentration (FIC) was obtained by dividing the colistin MIC in the presence of CA439 (0.312 μg mL−1) by the colistin MIC in the absence of CA439 (1.25 μg mL−1). Statistical analysis was performed using one-way ANOVA with Tukey’s correction multiple comparison test (**** P < 0.0001; ns, no significant differences).
CA439 effect on the PmrA/PmrB activity depends on PhoP/PhoQ integrity
PhoP/PhoQ and PmrA/PmrB TCS are functionally interconnected12 and they together activate the expression of genes that confer resistance to polymyxins. To assess whether PmrA/PmrB is directly targeted by CA439 and independently of PhoP/PhoQ, we analyzed ΔpmrA and double ΔphoP ΔpmrA mutant strains (Fig. 3). Both mutants behaved similarly to the ΔphoPQ strain (FIC of 0.25), consistent with the fact that PmrA/PmrB activity is subordinated to the PhoP/PhoQ TCS12.

CA439-colistin potentiation indirectly affects PmrA/PmrB system activity. Checkerboard assay showing CA439-colistin combination in (a) pmrA mutant or (b) phoP pmrA double mutant. Dark blue regions represent higher cell density. Data represent the mean OD (600 nm) of at least three biological replicates. Right panel shows the CFU counting obtained in each growth condition: DMSO (LB-medium with the addition of DMSO), CA439 (25 μM CA439 alone), colistin (0.156 μg mL−1 colistin alone) and CA439 + colistin (25 μM CA439 + 0.156 μg mL−1 colistin). Fractional inhibitory concentration (FIC) was obtained by dividing the colistin MIC in the presence of CA439 (0.156 μg mL−1) by the colistin MIC in the absence of CA439 (0.625 μg mL−1). Statistical analysis was performed using one-way ANOVA with Tukey’s correction multiple comparison test (**** P < 0.0001; ns, no significant differences).
To confirm that CA439 does not modulate PmrA/PmrB directly, we evaluated whether the compound could modulate the expression of L-Ara4N and PEtN modifications via PmrA/PmrB and independently of PhoP/PhoQ. arn or pmrCAB plasmidic transcriptional reporters were constructed by cloning the promoter regions of either operons upstream gfp that encodes the green fluorescent protein (GFP). The reporters were tested into the wild type, the ΔphoP or the ΔphoPQ mutant backgrounds, in the presence or absence of FeCl3 (activator of the PmrA/PmrB system)26, Mg2+ (repressor of the PhoP/PhoQ system)7 or CA439. Quinazoline CA439 inhibited the expression of the arn and the pmrCAB operons only in the presence of an intact phoPQ operon (Fig. 4a). No modulation of the fluorescence levels was detected in the ΔphoP or ΔphoPQ backgrounds, irrespective of the level of PmrA/PmrB induction achieved by addition of increasing concentrations of FeCl3 in the growth medium (Fig. 4b). These results rule out the direct effect of CA439 onto the PmrA/PmrB TCS system activity.

CA439 inhibitory effect on PmrA/PmrB activity depends on PhoP/PhoQ. (a) Transcriptional activity from arn::gfp (left) or pmrCAB::gfp (right) fusions in the wild type strain grown in the presence of 100 μM FeCl3, 5 mM MgCl2 or 25 μM CA439. (b) Transcriptional activity from pmrCAB::gfp fusion in a phoP (left) or phoPQ (right) mutant strain background grown in the presence of increasing concentrations of FeCl3 (25, 50, 75, 100 μM) with or without the addition of 25 μM CA439. Bacteria were grown at 37 °C for 16 h in the condition indicated in each assay. Transcriptional activity was calculated from the ratio between the fluorescence values (FU) and OD600 values (FU/OD600). The results represent the average of at least three independent experiments and error bars correspond to standard deviation (SD). Statistical analysis was performed using a, one-way ANOVA with Tukey’s correction multiple comparison test (****P < 0.0001, ***P < 0.001, **P < 0.01) or (b) unpaired t test (ns, no significant differences).
CA439-colistin potentiation phenotype partially depends on L-Ara4N and PEtN pathways
To explore whether CA439 might impact a PhoP/PhoQ and PmrA/PmrB independent pathway that modulates LPS modifications, we constructed mutant strains in arnA24 and pmrC, which disrupt L-Ara4N and PEtN lipid A modifications. Quinazoline CA439 combined with colistin was evaluated in a ΔarnA, ΔpmrC and ΔarnA ΔpmrC mutant backgrounds (Fig. 5). The inactivation of these two main LPS modification pathways was not sufficient to abrogate the enhancer phenotype, confirming that CA439 has an additional, PhoP/PhoQ-PmrA/PmrB-independent, effect.

L-Ara4N and PEtN pathways disruption does not completely reverse CA439-colistin potentiation. Checkerboard assay showing CA439-colistin combination in (a) arnA mutant, (b) pmrC mutant or c, arnA pmrC double mutant strains. Dark blue regions represent higher cell density. Data represent the mean OD (600 nm) of at least three biological replicates. Right panel shows the CFU counting obtained in each growth condition: DMSO (LB-medium with the addition of DMSO), CA439 (25 μM CA439 alone), colistin (0.078 μg mL−1 colistin alone) and CA439 + colistin (25 μM CA439 + 0.078 μg mL−1 colistin). Fractional inhibitory concentration (FIC) was calculated by dividing the colistin MIC in the presence of CA439 (0.078 μg mL−1) by the colistin MIC in the absence of CA439 (0.312 μg mL−1). Statistical analysis was performed using one-way ANOVA with Tukey’s correction multiple comparison test (****P < 0.0001; ns, no significant differences).
The CA439-colistin potentiation is partially Rcs-dependent
Membrane stress induced by CAMPs is also sensed by the Rcs TCS23,27. Rcs controls several cellular processes, among which is the synthesis of colanic acid (CA) that results in the production of capsular polysaccharide and the activation of ugd that is required for both CA biosynthesis and L-Ara4N modification of LPS23. To analyze this, we evaluated the effect of inactivating wcaL, wzxc, wzb or wcaJ, Rcs-dependent genes involved in CA synthesis28. Their inactivation did not alter the MIC value of colistin when compared with the wild type strain (Supplementary Figs. S3 and S4), indicating that the products of these genes have no role in the resistance mechanism.
We then analyzed the behavior of single ΔrcsB and a double ΔphoP ΔrcsB mutant strains in the presence of the CA439-colistin combination (Fig. 6). ΔrcsB partially reversed the phenotype (FIC of 0.25 compared to FIC of 0.125 for the wild type strain) (Fig. 6a) while the double mutant strain strongly attenuated the phenotype (Fig. 6b), indicating that there is at least one Rcs regulon member involved in CA439-colistin potentiation effect.

CA439-colistin potentiation is partially Rcs-dependent. Checkerboard assay showing CA439-colistin combination in (a) rcsB mutant or (b) phoP rcsB double mutant. Dark blue regions represent higher cell density. Data represent the mean OD (600 nm) of at least three biological replicates. Right panel in shows the CFU counting obtained in each growth condition: DMSO (LB-medium with the addition of DMSO), CA439 (25 μM CA439 alone), colistin (0.156 μg mL−1 for ΔrcsB and 0.078 μg mL−1 for ΔphoP ΔrcsB) and CA439 + colistin (25 μM CA439 + 0.156 μg mL−1 colistin for ΔrcsB and 0.078 μg mL−1 colistin for ΔphoP ΔrcsB). Fractional inhibitory concentration (FIC) was obtained by dividing the colistin MIC in the presence of CA439 (0.156 μg mL-1 for both strains) by the colistin MIC in the absence of CA439 (0.625 μg mL−1 for ΔrcsB and 0.156 μg mL−1 for ΔphoP ΔrcsB). Statistical analysis was performed using one-way ANOVA with Tukey’s correction multiple comparison test (****P < 0.0001; **P < 0.001; *P < 0.01; ns, no significant differences).
The transcriptional expression of the UDP-glucose dehydrogenase gene (ugd) required for both CA biosynthesis and L-Ara4N modification of LPS, depends on PhoP, PmrA and RcsB23. Therefore, a single Δugd and a double Δugd ΔpmrC mutant strains in which CA biosynthesis, L-Ara4N and PEtN modifications are affected were tested (Supplementary Fig. S5). The Δugd strain showed an eightfold decrease in MIC values and a FIC of 0.5 versus FIC of 0.125 for the wild type strain (Supplementary Fig. S5a). Δugd ΔpmrC and ΔphoP ΔrcsB strains behaved similarly (Supplementary Fig. S5b and Fig. 6b), indicating that CA439 inhibitory action impacts ugd and pmrC expression, increasing colistin sensitivity. However, a residual potentiation effect observed in ΔphoP ΔrcsB and Δugd ΔpmrC strains indicates that an additional mechanism still contributes to the CA439-dependent increased sensitivity to colistin.
CA439 selectively potentiates polymyxins
Next, we analyzed if the quinazoline could affect other bacterial targets different from colistin. We assayed CA439 combined with conventional antibiotics that act at peptidoglycan level (ampicillin, imipenem, meropenem), at protein translation level (chloramphenicol, kanamycin, azithromycin, tetracycline) or at gene transcription level (rifampicin). There was no alteration in the efficacy of antibiotics with mechanisms that do not involve LPS or the inner bacterial membrane (Supplementary Fig. S6), indicating that the potentiating effect of CA439 is selective for colistin (or for colistin-type compounds).
CA439 alters membrane polarization and permeabilizes the outer membrane
Therefore, we explored whether CA439 could exert an additional PhoP/PhoQ-PmrA/PmrB and Rcs-independent effect on the bacterial membrane.
The fluorescent probe DiSC3(5) is used to detect differences in membrane potential29. The probe is a positively charged and hydrophobic molecule that associates with polarized membranes. The IM of Gram-negative bacteria has a negative electrochemical potential, with accumulation of positive charges in the periplasmic space and negative charges in the intracellular space. On polarized bilayers, DiSC3(5) fluorescence is self-quenched as the molecule associates with the membrane. Alterations in membrane potential will make the probe increase its fluorescence as it migrates from the membrane29. Therefore, wild type bacteria were preincubated with the probe until stable basal fluorescence intensity was reached (5 min). After this time, colistin was added first (5 min) and increasing concentrations of either CA439, negative control CA490 or DMSO were subsequently added (Fig. 7a). CA439 promoted a decrease in fluorescence intensity to basal levels, indicating entry of the probe. This shows an effect at the IM level. Consistent with the CA439-specific phenotypes obtained so far, CA490 caused no alteration.

CA439 alters IM potential and promotes colistin-mediated lysis. (a) Fluorescence intensity was monitored from a wild type strain culture incubated with 1 μM DiSC3(5) for 5 min and then with 5 μg mL−1 colistin for another 10 min followed by the indicated concentration of CA439, 25 μM CA490 or the equivalent volume of DMSO used for the maximum concentration of compound tested. Statistical analysis was performed using one-way ANOVA with Tukey’s correction multiple comparison test against no addition condition (-) (** P < 0.001; * P < 0.01; ns, no significant differences). (b) OM disruption and c, IM disruption were measured using β-lactamase assay and β-galactosidase assay, respectively, as described in Materials and Methods. Cells expressing (b) β-lactamase or (c) β-galactosidase were incubated with PBS (Saline), DMSO or increasing concentrations of CA439 (left panel) or colistin (b) from 0 to 2.5 μg mL−1 or (c) from 0 to 200 μg mL−1) in the presence of 25 μM CA439 (grey bars) or DMSO (dark bars) (right panel). (b) Nitrocefin (30 μM) or (c) ONPG (2 mM) were added as substrate and absorbance at 492 nm and 405 nm, respectively, were monitored. Statistical analysis was performed using unpaired t test (**** P < 0.0001; *** P < 0.001; ns, no significant differences). The results are the average of at least three independent experiments.
Next, we compared the permeabilization action of colistin and/or CA439 on the bacterial IM versus the OM. We tested the ability of the compounds to allow the entry of two substrates that require a previous permeabilization of the OM or the IM to get in contact with a hydrolytic enzyme: nitrocefin, substrate of periplasmic beta-lactamase, or o-nitrophenyl-β-galactoside (ONPG), substrate of cytoplasmic beta-galactosidase. While colistin is able to alter both IM and OM integrity30, CA439 alone did not disrupt IM integrity and it did not alter OM permeability unless it was combined with colistin, enhancing its effect (Fig. 7b,c).
Together, the results indicate that CA439 promotes the entry of colistin through the OM and exerts an effect on the IM by altering proton motive force homeostasis.
CA439 shows effectiveness against strains with high resistance to colistin
Resistance to colistin mainly occurs by the pmrC, ugd and the arn-mediated modification of LPS. However, two main acquired resistance mechanisms to colistin in Enterobacteriaceae have been described. Mutations or disruptions of mgrB, which encodes for MgrB, a transmembrane peptide that inhibits the PhoP/PhoQ system, derepress the system and thus increase resistance to polymyxins31. The plasmid-mediated colistin resistance (Mcr) is easily transferred among bacteria and confers resistance to colistin-susceptible strains. mcr-1 encodes for the most frequent phosphoethanolamine (PEtN) transferase enzyme variant identified in resistant clinical isolates. MCR-1 incorporates PEtN into lipid A similar to the pmrC gene product32. Therefore, CA439-colistin effect was analyzed in a ΔmgrB mutant strain and in a wild type strain that overexpresses mcr-132 (Fig. 8). In either case, CA439 caused a reduction in the MIC of colistin. As expected, a ΔphoPQ ΔmgrB double mutant strain rendered an equivalent MIC for colistin and FIC value to the single ΔphoPQ strain (Supplementary Fig. S7).

CA439 is effective against highly resistant colistin strains. Checkerboard assay showing CA439-colistin combination in (a) a mgrB mutant or (b) in a wild type strain overexpressing mcr-1. Dark blue regions represent higher cell density. Data represent the mean OD (600 nm) of at least three biological replicates. Right panel shows the CFU counting obtained in each growth condition: DMSO (LB-medium with the addition of DMSO), CA439 (25 μM CA439 alone), colistin (1.25 μg mL−1 and 2.5 μg mL−1 colistin alone for ΔmgrB and mcr-1 respectively) and CA439 + colistin (25 μM CA439 + 1.25 μg mL −1 colistin for ΔmgrB or 2.5 μg mL−1 colistin for mcr-1). Fractional inhibitory concentration (FIC) was obtained by dividing the colistin MIC in the presence of CA439 (1.25 μg mL−1 for ΔmgrB and 2.5 μg mL−1 for mcr-1) by the colistin MIC in the absence of CA439 (20 μg mL-1 for both strains). Statistical analysis was performed using one-way ANOVA with Tukey’s correction multiple comparison test (****P < 0.0001; ***P < 0.001; **P < 0.01; ns, no significant differences).
We evaluated whether we could extend the use of CA439 to other Salmonella strains that are resistant to colistin (S. Typhimurium STM23 and S. Enteritidis PT4) and in other bacterial genera of clinical interest (E. coli, A. baumannii and K. aerogenes). Although both Salmonella strains show a fourfold increase in colistin resistance (MIC of 2.5 μg mL−1 for 14028 s compared to 20 μg mL−1 for STM23 and PT4), CA439 was able to potentiate colistin, reaching FIC values of 0.03125 and 0.0625 for STM23 and PT4, respectively (Supplementary Fig. S8). E. coli strains BW25113 and MG1655 (FIC of 0.25), A. baumannii (FIC of 0.125) and K. aerogenes (FIC of 0.5) also presented enhanced susceptibility to the combined drugs when compared with the effect exerted by colistin alone (Supplementary Fig. S9).
CA439 potentiates colistin-mediated lysis
We then evaluated whether CA439 was capable of altering colistin-mediated lysis of Salmonella. Either the wild type or ΔmgrB strains were preincubated with CA439 for 3 h, followed by treatment with a lytical concentration of colistin and the optical density of the cultures was monitored. Preincubation with CA439 favored bacterial cell lysis promoted by colistin (black curves) while the addition of DMSO had no effect (Supplementary Fig. S10, black curves). To analyze whether preincubation with CA439 was required, the quinazoline was added individually or simultaneously with colistin at 3 h (Supplementary Fig. S10, blue curves). Either protocol showed equivalent effects (Supplementary Fig. S10), indicating that preincubation was not a prerequisite. These results reinforce the notion that CA439 aids in destabilizing the bacterial membrane (Fig. 7).
Colistin-CA439 protective effect in the Galleria mellonella infection model
To examine if the CA439-colistin combination could be used as an efficient therapy in vivo, G. mellonella was chosen as a suitable infection model33. No toxic effects were observed below 5 mg kg−1 and 25 mg kg−1 of colistin and CA439, respectively (Supplementary Fig. S11, left and middle panel). Because the combination of drugs showed toxic effects up to 0.25 mg kg−1 (Supplementary Fig. S11, right panel), we used 0.125 mg kg−1 (0.0625 mg kg−1 of each compound). Larvae groups were infected with 1 × 107 CFU/larva of Salmonella. After 2 h, larvae were treated with colistin or CA439 alone or with the combination of drugs and survival was monitored up to 72 h post-infection. The results indicate that while each individual drug did not show significant difference when compared to the injection of PBS (control) up to 72 h post-infection, the treatment with the CA439 and colistin combination of the drugs showed 30 and 33% increase in survival of the larvae at 48 and 72 h post-infection, respectively (Fig. S12). As expected, the ΔphoP strain used as control was strongly attenuated in its virulence ability when compared to the wild type strain (Fig. S12).
Discussion
Colistin, a polymyxin chemotype drug, is currently used as a last-resort therapy for bacterial infections caused by multidrug-resistant pathogens, such as Salmonella enterica, Acinetobacter baumannii and Pseudomonas aeruginosa. However, colistin doses applied with effective antimicrobial action are associated with significant toxic adverse effects so its use is carefully considered17,31.
In this study, we aimed to find a novel potential therapeutic approach for the treatment of salmonellosis, taking into account the mechanisms that allow this pathogen to counteract the effect of colistin. One key player is the Salmonella PhoP/PhoQ TCS which, upon interaction with polymyxins upregulates the expression of a set of genes including those that mediate the attachment of aminoarabinose to lipid A and the modification of phosphate groups (particularly with phospho-ethanolamine) in the LPS core region24. Such alterations diminish the charge density associated with LPS, thereby decreasing the affinity of the cationic colistin molecule for the bacterial surface. To attain a reduction in the effective antibacterial colistin concentration, we combined this knowledge with our previous identification of quinazoline molecules which selectively inhibit PhoQ autophosphorylation by competition with ATP16. We demonstrate that CA439 quinazoline lowers the MIC of colistin with a fractional inhibitory concentration (FIC) of 0.125. We compared the phenotypes of the wild type with single and double mutants that inactivate the main PhoP/PhoQ-dependent genes responsible for CAMPs resistance, including phoP, phoQ, and PhoP-indirectly regulated- PmrA/PmrB-dependent genes, pmrC and the arn operon (pmrHIJKLM). In all cases, the reduction in the FIC values relied on the inhibitory effect of the quinazoline on the activity of the PhoP/PhoQ system as a master regulator of the cascade. However, the inactivation of none of the aforementioned genes, including either phoP or phoQ, was able to completely abrogate the enhancing action of the quinazoline.
We determined that an ugd (also named pmrE) mutant strain was eightfold more susceptible to colistin than the wild type one, indicating that this gene product confers a layer of resistance beyond other PhoP/PhoQ-dependent genes. The ugd gene encodes the UDP-glucose dehydrogenase enzyme that converts UDP-glucose in UDP-glucuronic acid, the precursor molecule of synthetic pathways that render extracellular polysaccharides (including CA), and L-Ara4N, both factors required to counteract polymyxin deleterious action24.
ugd transcription depends on the concert of PhoP, PmrA and Rcs regulators, which directly bind to ugd promoter region, being PhoP required to activate ugd expression when bacteria are under a low magnesium, low Fe3+ concentration environment (in which PmrA is activated via the PhoP-PmrD cascade)12. In this condition, a double pmrC (deficient in PEtN modification in LPS) and ugd mutant strain (deficient in exopolysaccharides and L-Ara4N synthesis) showed a reduction in colistin MIC value by 16-fold when compared to the wild type one. Intriguingly, the single inactivation of main capsule synthetic genes, wcaL, wzxc, wzb and wcaJ, all known to be under Rcs regulation28, did not significantly alter neither colistin MIC levels nor the combined quinazoline-colistin effect. Therefore, the Rcs-dependent gene that plays a key role in the contribution to colistin resistance is ugd, mainly by its involvement in the synthesis of the UDP-glucuronic acid, the precursor of the pathway that renders L-Ara4N-modified LPS. Still, in this context, when the major described pathways that confer resistance cannot be activated (i.e., in the phoP rcsB double mutant strain), a remnant potentiation of colistin action by the quinazoline could be observed.
The quinazoline CA439 was unable to enhance the action of antibiotics that target bacterial pathways that differ from colistin, such as DNA transcription, RNA translation or cell wall peptidoglycan synthesis. These results indicate that the depolarization action of CA439 is not enough to exert a potentiating effect when combined with other antimicrobial agents distinct from colistin. Moreover, the quinazoline did not enhance the sub-lytic bacterial permeabilizing action of EDTA (a cation chelating drug) or SDS (anionic detergent). However, in addition to the inhibitory action over the PhoQ regulon, CA439 was effective in disrupting Salmonella OM electrochemical potential, that together with the action of colistin produces the collapse of its homeostatic mechanisms. Whilst it is well described that once polymyxins traverse the OM, they permeabilize the IM, which is required for bacterial lysis34, the mechanistic details of IM disruption have not been entirely elucidated. Recent work indicates that, once colistin reaches the periplasm, it can bind a pool of precursor LPS molecules that are associated to the IM and waiting to be transported to the outer membrane leaflet, and that the induced accumulation of LPS in the IM augments the bacterial susceptibility to colistin19. The fact that CA439 does not alter the IM integrity was also strengthen by its incapacity to enable the access of ONPG to be hydrolyzed by the cytoplasmic β-galactosidase enzyme. In contrast, and reinforcing the electrochemical imbalance results, only when it acts in conjunction with colistin, the quinazoline allows the entry of nitrocefin to be hydrolyzed by periplasmic β-lactamase. So, it is tempting to postulate that the electrochemical OM disturbance caused by the quinazoline facilitates the intrinsic capacity of colistin to disorganize the OM LPS, an effect that subsequently facilitates a better access to perturb the integrity of the cytoplasmic membrane. We also show that the dual drug treatment improved the lytic performance of colistin when compared treatment with an equivalent, sub-lytic concentration of colistin alone (the cartoon in Fig. S13 depicts our model for the concerted action of CA439).
One major drawback for the success of colistin antibacterial therapy, in addition to toxic liabilities over the host, is the evolving bacterial resistance due to acquired mutations in key components of the PhoP/PhoQ regulon17,35,36. Among PhoQ-regulating proteins, MgrB is the most widely distributed among Enterobacteriaceae37 Being a repressor of PhoQ, mgrB loss of function mutants exacerbate PhoQ-dependent resistance against colistin. In Klebsiella pneumoniae or in Pseudomonas aeruginosa, mutations or disruption of the mgrB gene have been shown to be prevalent among other chromosomal mutations in colistin-resistant clinical strains38. When CA439 is added, a Salmonella mgrB mutant showed a 16-fold decrease in colistin MIC values, pointing at the efficacy of the dual action of the compounds even when the PhoP/PhoQ system is derepressed. S. Typhimurium STM23 and S. enteritidis PT4 clinical isolates39,40 showed 16 to 32-fold increase in susceptibility to colistin when they were under the dual drug effect, demonstrating that the potentiating effect can be applied to strains with clinical impact, other than S. enterica serovar Typhimurium 14028 s.
A Salmonella strain which expresses mcr-1, one of the most extensively characterized and widespread mobilizable plasmid-borne resistance genes, assists in dissemination of colistin resistance to other pathogenic bacteria by horizontal transfer32, displayed eightfold reduction of the resistance to colistin when exposed to the combined treatment. In this case, the capacity of the mcr-1 encoded enzyme in modifying LPS with PEtN residues would be insufficient to confer resistance when PhoP/PhoQ- and Rcs-dependent determinants for resistance have been inhibited by the quinazoline action. The combined antibacterial treatment was also more effective in E. coli, A. baumannii and Klebsiella aerogenes strains when compared with colistin alone. This correlates with the impact that conserved PhoP/PhoQ-PmrA/PmrB and Rcs-dependent mechanisms have in polymyxin resistance in these Gram-negative pathogens and this would extend the prospective application of the mixed treatment.
The clinical reintroduction of colistin, combined with the extensive use for veterinary medicine, increases the prevalence of resistant strains in livestock and the concomitant risk of animal-human transmission17. In this context, our results foresee an auspicious therapeutic outcome for the dual agent strategy when applied to treat highly resistant, clinically hazardous strains.
The combination of colistin and quinazoline mitigated Salmonella’s lethal effects in a systemic infection model using Galleria mellonella wax moth larvae when compared to the administered sub-lethal concentration of each single-drug. Overall, our results encourage further pursuit of the therapeutic implementation of the combined quinazoline-colistin treatment in mammalian hosts to combat Salmonella infections.
Materials and methods
Bacterial strains, cell culture and growth conditions
A list of the strains and plasmids used in this work is provided in Supplementary Table S1. Bacteria were routinely grown in Miller’s Luria–Bertani (LB) medium or on LB agar plates at 37 °C supplemented with appropriated antibiotics (kanamycin, 50 μg mL−1; chloramphenicol, 20 μg mL−1; ampicillin, 100 μg mL−1; tetracycline, 12.5 μg mL−1; spectinomycin, 50 μg mL−1).
Strains carrying gene deletions or chromosomal lacZ reporter fusions were generated by Lamba Red-mediated recombination followed by P22-mediated transduction using previously described protocols41. Insertion mutations in pmrA, arnA and pmrC were constructed with the one-step chromosomal inactivation method42. The promote regions of arn and pmrCAB operons were amplified by PCR and ligated into the pPROBE(NT) plasmid43. Primers used are indicated in Supplementary Table S2.
All reagents and chemicals were from Sigma, except the Luria–Bertani culture medium that was from Difco. Colistin was obtained from Sigma (Colistin sulfate salt, Cat#4461). Oligonucleotides and enzymes were purchased from Life Technologies. SMILES and Labbook codes for the compounds are provided in Supplementary Table S3 and they were synthesized as previously described16.
Checkerboard Assay
Overnight cultures (18 h) of each strain grown in LB medium were sub-cultured 1:10.000 in fresh LB medium supplemented with CA439 (or CA439-analogues) and added to a 96-well assay plate containing the indicated twofold dilutions of colistin (EDTA or SDS when appropriate). Bacteria were grown at 37 °C with shaking at 250 rpm to stationary phase and OD600 was measured using a Synergy 2 plate reader. OD600 data was converted to percent growth and heatmaps were generated in GraphPad (version 6.01; GraphPad Software, San Diego, CA, USA), with a scale between 0–100% growth in which dark blue regions represent higher cell density and light blue regions lower cell density. Raw data for each strain assayed is provided in Supplementary Table S4. The MIC values were determined to be the condition that resulted in a percent residual growth of ≤ 10%. The FIC was determined to be the MIC of colistin combined with the indicated concentration of the compound divided by the MIC of colistin alone. Overnight cultures were used for serial dilutions to determine the colony-forming units (CFU) in each condition. CFU evaluations were carried out with individual sub-inhibitory concentrations of CA439 or of colistin, or with the combination of these concentrations of drugs, as indicated in the legend of the corresponding figures.
β-galactosidase activity assays
Bacteria were grown overnight with shaking at 37 °C in LB media with the addition of CA439, CA490, colistin or colistin combined with CA439 or CA490 at the final concentration indicated in each experiment. Kanamycin (50 μg mL−1) or chloramphenicol (20 μg mL−1) were added when required. β-galactosidase activity was determined as described44. Relative activity was calculated taking the activity obtained in DMSO as 100%.
GFP fluorescence assays
The S. Typhimurium wild type strain carrying the pParn-gfp or pPpmrCAB-gfp reporter plasmid or ΔphoP and ΔphoPQ strains carrying the pPpmrCAB-gfp reporter plasmid were grown with shaking overnight at 37 °C. Cultures were subcultured 1:100 in LB medium and supplemented with FeCl3 (100 μM), MgCl2 (5 mM) or CA439 (25 μM) for the wild type strain or with the indicated concentration of FeCl3 and CA439 (25 μM) or the equivalent of DMSO for ΔphoP and ΔphoPQ strains and were incubated at 37 °C with shaking for 16 h. Optical density at 600 nm (OD600) and GFP fluorescence (excitation at 485/emission at 528 nm) were determined in a microwell plate reader (Synergy 2). Transcriptional activity was calculated as the ratio of GFP fluorescence and OD600 (FU/OD600). Relative FU/DO600 was calculated by taking the activity obtained in DMSO as 100%.
DiSC 3(5) assay
Cells were grown to early-exponential phase in LB media supplemented with 5 mM EDTA to disrupt the outer membrane and promote DiSC3(5) access to the cytoplasmic membrane. Cells were harvested, washed twice with 5 mM Bis–Tris/Trizma, 20 mM glucose pH 7, and resuspended in the same buffer to a final OD600 of 0.1 with 1 μM DiSC3(5). Cells were transferred to a gently stirred cuvette at 37 °C and DiSC3(5) fluorescence (excitation = 620 nm, emission = 670 nm) was read immediately after using a Cary Eclipse Fluorescence Spectrophotometer (Agilent). After steady-state fluorescence was reached (5 min), 5 μg mL-1 colistin was added (5 min) and then the indicated concentration of CA439 (CA490 or the equivalent of DMSO) was added. The fluorescence intensity was recorded for at least 15 min after compound addition.
Outer membrane integrity assay
The β-lactamase assay was performed as previously described45,46. S. Typhimurium 14028 s pBR322 cells grown overnight in LB medium with 100 μg mL−1 ampicillin were subcultured 1:50 in fresh LB with 50 μg mL−1 ampicillin and grown at 37 °C to early-exponential phase. Cells were harvested, washed in PBS and resuspended to OD600 = 0.02. A volume of 50 μL of the cell suspension was added to a 96-well plate containing 50 μL of PBS with a final concentration of 30 μM nitrocefin and the antibiotic, the compound or the combination indicated in each assay. Plates were incubated at 37 °C and absorbance at 492 nm was read after 2 h using a microwell plate reader (Synergy 2) to monitor nitrocefin hydrolysis.
Inner Membrane Integrity Assay
The β-galactosidase assay was performed as previously described45,46 .S. Typhimurium 14028 s cpxP::lacZ cells grown overnight in LB medium, subcultured 1:50 in fresh LB and grown at 37 °C to early-exponential phase. Cells were harvested, washed in PBS and resuspended to OD600 = 0.4. A volume of 50 μL of the cell suspension was added to a 96-well plate containing 50 μL of PBS with a final concentration of 2 mM o-nitrophenyl-β-galactoside (ONPG) and the antibiotic, the compound or the combination indicated in each assay. Plates were incubated at 37 °C and absorbance at 405 nm was read after 1 h using a microwell plate reader (Synergy 2) to monitor ONPG hydrolysis.
Colistin-mediated lysis curves
Overnight cultures (18 h) grown in LB medium were subcultured 1:100 in fresh LB medium supplemented with CA439, CA490 or the equivalent of DMSO (black curves). After 3 h, a concentration of colistin equivalent to 6xMIC (for wild type) or 1xMIC (for ΔmgrB) was added alone or combined with CA439 or CA490. Bacteria were grown at 37 °C with shaking at 200 rpm and OD600 was measured every hour. After 8 h, serial dilutions of the cultures were made for colony-forming units’ determination.
G. mellonella infection model
G. mellonella survival assays were performed as described previously47,48. Briefly, 16 larvae per group were selected by their weight, variable between 0.18 and 0.3 g, and incubated overnight at 37 °C without food. Each group was inoculated with bacterial suspensions prepared in PBS buffer (saline) of the strains under study at the CFU/larva values mentioned in the text. The inoculum was prepared at a fixed concentration, determined by plate count, in a PBS buffer and 10% glycerol, frozen and then diluted to obtain the necessary concentration to inoculate49. The chosen inoculation site was the last left proleg. The compounds analyzed were administered to the right proleg 2 h after inoculation with the bacteria. For the survival curves, the status of the larvae was checked every 24 up to 72 h post-infection.
Statistical analysis
To test for statistical differences between means, one-way analysis of variance (ANOVA) and the Tukey multiple comparison test with an overall significance level of 0.05 were used. Calculations were performed with GraphPad Prism statistical software. When required, unpaired t test was used with the same overall significance level. Kaplan–Meier curves were constructed to analyze the toxic effect of the compounds over Galleria larvae50, while two-way ANOVA statistical analysis was used to analyze the Galleria larvae survival assay.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Ventola, C. L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 40, 277–283 (2015).
Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 399, 629–655 (2022).
Theuretzbacher, U., Outterson, K., Engel, A. & Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 18, 275–285 (2020).
De Cesare, A. Salmonella in foods: A reemerging problem. Adv. Food Nutr. Res. 86, 137–179 (2018).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
Castelli, M. E., García Véscovi, E. & Soncini, F. C. The phosphatase activity is the target for Mg2+ regulation of the sensor protein PhoQ in Salmonella. J. Biol. Chem. 275, 22948–22954 (2000).
García Véscovi, E., Soncini, F. C. & Groisman, E. A. Mg2+ as an extracellular signal: Environmental regulation of Salmonella virulence. Cell 84, 165–174 (1996).
Prost, L. R. et al. Activation of the bacterial sensor kinase PhoQ by acidic pH. Mol. Cell 26, 165–174 (2007).
Bader, M. W. et al. Regulation of Salmonella typhimurium virulence gene expression by cationic antimicrobial peptides. Mol. Microbiol. 50, 219–230 (2003).
Soncini, F. C., García Véscovi, E., Solomon, F. & Groisman, E. A. Molecular basis of the magnesium deprivation response in Salmonella typhimurium: Identification of PhoP-regulated genes. J. Bacteriol. 178, 5092–5099 (1996).
Groisman, E. A. et al. Bacterial Mg2+ homeostasis, transport, and virulence. Annu. Rev. Genet. 47, 625–646 (2013).
Chen, H. D. & Groisman, E. A. The biology of the PmrA/PmrB two-component system: the major regulator of lipopolysaccharide modifications. Annu. Rev. Microbiol. 67, 83–112 (2013).
Bearson, B. L., Wilson, L. & Foster, J. W. A low pH-inducible, PhoPQ-dependent acid tolerance response protects Salmonella typhimurium against inorganic acid stress. J. Bacteriol. 180, 2409–2417 (1998).
Ernst, R. K., Guina, T. & Miller, S. I. How intracellular bacteria survive: Surface modifications that promote resistance to host innate immune responses. J. Infect. Dis. 179(Suppl 2), S326–S330 (1999).
Calvert, M. B., Jumde, V. R. & Titz, A. Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections. Beilstein J. Org. Chem. 14, 2607–2617 (2018).
Carabajal, M. A. et al. Quinazoline-Based Antivirulence Compounds Selectively Target Salmonella PhoP/PhoQ Signal Transduction System. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.01744-19 (2019).
Andrade, F. F., Silva, D., Rodrigues, A. & Pina-Vaz, C. Colistin update on its mechanism of action and resistance, present and future challenges. Microorganisms 8(11), 1716 (2020).
Velkov, T., Thompson, P. E., Nation, R. L. & Li, J. Structure–activity relationships of polymyxin antibiotics. J. Med. Chem. 53, 1898–1916 (2010).
Sabnis, A. et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. Elife https://doi.org/10.7554/eLife.65836 (2021).
Vázquez, X. et al. Colistin resistance in monophasic isolates of Salmonella enterica ST34 collected from meat-derived products in Spain, with or without CMY-2 Co-production. Front. Microbiol. https://doi.org/10.3389/fmicb.2021.735364 (2022).
Soncini, F. C. & Groisman, E. A. Two-component regulatory systems can interact to process multiple environmental signals. J. Bacteriol. 178, 6796–6801 (1996).
Bertani, B. & Ruiz, N. Function and biogenesis of lipopolysaccharides. EcoSal Plus https://doi.org/10.1128/ecosalplus.esp-0001-2018 (2018).
Mouslim, C. & Groisman, E. A. Control of the Salmonella ugd gene by three two-component regulatory systems. Mol. Microbiol. 47, 335–344 (2003).
Raetz, C. R. H., Reynolds, C. M., Trent, M. S. & Bishop, R. E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 (2007).
Tyers, M. & Wright, G. D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 17, 141–155 (2019).
Wösten, M. M., Kox, L. F., Chamnongpol, S., Soncini, F. C. & Groisman, E. A. A signal transduction system that responds to extracellular iron. Cell 103, 113–125 (2000).
Farris, C., Sanowar, S., Bader, M. W., Pfuetzner, R. & Miller, S. I. Antimicrobial peptides activate the Rcs regulon through the outer membrane lipoprotein RcsF. J. Bacteriol. 192, 4894–4903 (2010).
Meng, J., Young, G. & Chen, J. The RCS system in Enterobacteriaceae: Envelope stress responses and virulence regulation. Front. Microbiol. 12, 627104 (2021).
Te Winkel, J. D., Gray, D. A., Seistrup, K. H., Hamoen, L. W. & Strahl, H. Analysis of antimicrobial-triggered membrane depolarization using voltage sensitive dyes. Front. Cell Dev. Biol. 4, 29 (2016).
Klobucar, K. et al. Genetic and chemical screening reveals targets and compounds to potentiate gram-positive antibiotics against gram-negative bacteria. ACS Infect. Dis. 8, 2187–2197 (2022).
Aghapour, Z. et al. Molecular mechanisms related to colistin resistance in Enterobacteriaceae. Infect. Drug Resist. 12, 965–975 (2019).
Ling, Z. et al. Epidemiology of mobile colistin resistance genes mcr-1 to mcr-9. J. Antimicrob. Chemother. 75, 3087–3095 (2020).
Bender, J. K., Wille, T., Blank, K., Lange, A. & Gerlach, R. G. LPS structure and PhoQ activity are important for Salmonella Typhimurium virulence in the Galleria mellonella infection model [corrected]. PLoS ONE 8, e73287 (2013).
Garg, S. K. et al. Resurgence of polymyxin B for MDR/XDR gram-negative infections: An overview of current evidence. Crit. Care Res. Pract. 2017, 3635609 (2017).
Olaitan, A. O., Morand, S. & Rolain, J.-M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 5, 643 (2014).
Poirel, L. et al. The mgrB gene as a key target for acquired resistance to colistin in Klebsiella pneumoniae. J. Antimicrob. Chemother. 70, 75–80 (2015).
Yadavalli, S. S. et al. Functional determinants of a small protein controlling a broadly conserved bacterial sensor kinase. J. Bacteriol. https://doi.org/10.1128/JB.00305-20 (2020).
Shein, A. M. S. et al. High prevalence of mgrB-mediated colistin resistance among carbapenem-resistant Klebsiella pneumoniae is associated with biofilm formation, and can be overcome by colistin-EDTA combination therapy. Sci. Rep. 12, 12939 (2022).
Thomson, N. R. et al. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome. Res. 18, 1624–1637 (2008).
Cordeiro, N. F. et al. Identification of the first blaCMY-2 gene in Salmonella enterica serovar Typhimurium isolates obtained from cases of paediatric diarrhoea illness detected in South America. J. Glob. Antimicrob. Resist. 1, 143–148 (2013).
Viarengo, G. et al. Unsaturated long chain free fatty acids are input signals of the Salmonella enterica PhoP/PhoQ regulatory system. J. Biol. Chem. 288, 22346–22358 (2013).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645 (2000).
Miller, W. G., Leveau, J. H. & Lindow, S. E. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant Microbe. Interact. 13, 1243–1250 (2000).
Griffith, K. L. & Wolf, R. E. Measuring beta-galactosidase activity in bacteria: Cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 290, 397–402 (2002).
Klobucar, K. et al. Chemical screen for vancomycin antagonism uncovers probes of the gram-negative outer membrane. ACS Chem. Biol. 16, 929–942 (2021).
Farha, M. A. et al. Uncovering the hidden antibiotic potential of Cannabis. ACS Infect. Dis. 6, 338–346 (2020).
Martino, G. P., Perez, C. E., Magni, C. & Blancato, V. S. Implications of the expression of Enterococcus faecalis citrate fermentation genes during infection. PLoS ONE 13, e0205787 (2018).
Martino, G. P. et al. Safety assessment and functional properties of four enterococci strains isolated from regional Argentinean cheese. Int. J. Food Microbiol. 277, 1–9 (2018).
Terán, L. C. et al. Genomic analysis revealed conserved acid tolerance mechanisms from native micro-organisms in fermented feed. J. Appl. Microbiol. 132, 1152–1165 (2022).
Rich, J. T. et al. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Head Neck Surg. 143, 331–336 (2010).
Acknowledgements
We thank Marina Avecilla for excellent technical assistance, Fernando C. Soncini for useful suggestions and Alejandro J. Vila and Dario Krapf for the kind gift of reagents. E.G.V., V.S.B. and C.M. are Career Investigators of Consejo de Investigaciones Científicas y Tecnológicas (CONICET), Argentina. This work was partially supported by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT 2020-02620 to E.G.V., PICT 2018-3932 to V.S.B.) and by CONICET (CONICET-22920160100039CO). Ph.D. studentships support C.A.L. (CONICET) and F.O.G. (ANPCyT).
Author information
Authors and Affiliations
Contributions
E.G.V. conceived the project. C.A.L. generated preliminary data that influenced the conception of the project. E.G.V. and C.A.L. generated hypotheses and designed the experiments. C.A.L. performed all the experiments. C.R.M.A. synthesized the quinazoline compounds. F.O.G., V.S.B., C.M., L.Y., A.R. and J.A.C. provided set up, resources, facility, protocol design and execution of in vivo assays. E.G.V. and C.A.L. analyzed the data and wrote the manuscript. All authors contributed to critical discussions and review of the manuscript. E.G.V. acquired funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lobertti, C.A., Gizzi, F.O., Magni, C. et al. Enhancing colistin efficacy against Salmonella infections with a quinazoline-based dual therapeutic strategy. Sci Rep 14, 5148 (2024). https://doi.org/10.1038/s41598-024-55793-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55793-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
