
Abstract
The oral and gastrointestinal mucosae represent the main targets of the toxic effect of chemo and/or radiotherapy administered during the conditioning regimen before hematopoietic stem cell transplant (HSCT). These harmful consequences and the immunological complications that may occur after the transplant (such as Graft versus Host Disease, GvHD) are responsible for the clinical symptoms associated with mucositis during the aplasia phase, like pain, nausea, vomiting, and diarrhea. These toxicities could play a critical role in the oral and gastrointestinal microbiomes during the post-transplant phase, and the degree of microbial dysbiosis and dysregulation among different bacterial species could also be crucial in intestinal mucosa homeostasis, altering the host’s innate and adaptive immune responses and favoring abnormal immune responses responsible for the occurrence of GvHD. This prospective pediatric study aims to analyze longitudinally oral and gut microbiomes in 17 pediatric patients who received allogeneic HSCT for malignant and non-malignant diseases. The oral mucositis was mainly associated with an increased relative abundance of Fusobacteria, and Prevotella species, while Streptococcus descendants showed a negative correlation. The fecal microbiome of subjects affected by cutaneous acute GvHD (aGvHD) correlated with Proteobacteria. Oral mucosal microbiota undergoes changes after HSCT, Fusobacteria, and Prevotella represent bacterial species associated with mucositis and they could be the target for future therapeutic approaches, while fecal microbiome in patients with acute GvHD (aGvHD) revealed an increase of different class of Proteobacteria (Alphaproteobacteria and Deltaproteobacteria) and a negative correlation with the class of Gammaproteobacteria.
Similar content being viewed by others


Introduction
The oral bacterial ecosystem represents a complex equilibrium frequently challenged by habits such as poor oral hygiene as well as the use of antimicrobial agents, physiological hormonal changes, age and dental development, and external pathogens. When this balance is lost, dysbiosis occurs with pathological complications1. The microbiota plays a crucial role in regulating host-microbe interactions among bacterial species, and its sensitivity to medical treatment can influence inflammation and the breakdown of the mucosal barrier2,3. In the context of hematopoietic stem cell transplantation (HSCT), both the oral and gut microbiota can become dysbiotic due to various factors that can affect the intestinal ecosystem and cause damage to both the oral and intestinal mucosa. These factors can include the use of antibiotics, chemotherapeutic drugs, alloimmune reactions like GvHD, and prolonged inflammatory states4,5. Oral mucositis (OM) represents one of the most frequent clinical complications observed in patients who received chemotherapy for the treatment of malignant diseases or during the conditioning regimen (CR) before HSCT6,7,8. Myeloablative regimens (MAC) are more frequently associated with severe toxic complications, including oral and gastro-intestinal mucositis, compared to reduced intensity conditioning regimens (RIC), and it is recognized that the development of severe mucositis is correlated with the performance status at transplant and with the previous toxicity occurred before the HSCT3. Patients with OM could develop oral ulceration associated with intense pain that compromises oral intake, and even lead to bloodstream infections, resulting in a decreased patient’s quality of life, prolonged hospitalization, and high treatment costs. The occurrence of infection starting from the oral cavity is facilitated by the post-transplant neutropenia phase9. The increased possibility of the risk of infection in transplanted patients is related to the immunosuppressive therapy administered to prevent and treat GvHD that represents “per se” the higher cause of immunodepression. The relationship between acute toxicity and the occurrence of GvHD is known, and who received MAC have a higher risk to develop acute toxicity compared to those who received RIC. Moreover, gastrointestinal GvHD induces a loss of intestinal flora diversity and alteration of microbiota10.
This study aims to analyze the oral and intestinal microbiome in a cohort of pediatric patients who received allogeneic HSCT and to describe the microbiological characteristic of their microbiome in the different phases of transplant and in patients who developed oral/gastrointestinal (GI) mucositis and acute GvHD.
Methods
Study design
For this prospective single-center study, we included 17 patients who received an allogeneic HSCT at IRCCS Istituto G. Gaslini from 2019 to 2022. The following variables: gender, age, underlying disease, oral and GI mucositis and its grade, type of CR, GvHD prophylaxis, and acute and chronic GvHD were included in the analysis. A prospective daily patient oral assessment was recorded, including diet, pain, erythema, ulceration, and the oral mucositis grade based on the World Health Organization (WHO) grading scale. Patient with acute gastrointestinal toxicity was recorded using Common Terminology Criteria of Adverse Events (CTCAE) grading. Acute and chronic GvHD was assessed using the standard criteria11,12. Samples for oral and fecal microbiome analysis were collected before HSCT, at engraftment, and 30 or 100 days after transplant. All patients gave their informed consent to participate in the study.
The study was conducted following the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Regione Liguria 369/2018.
Sample collection
The specimens were sampled in the morning without the patient having drunk, or eaten anything, performed oral hygiene in the previous half hour, or used mouthwashes or oral antiseptics, which would affect the result of the microbiological analyses. The swab was placed at the level of the mouth vestibule of the lower arch and rubbed about ten times along the entire length of each gingival semi-arch. At the end of the collection, the swab was repositioned inside the tube containing a stabilizing solution and vigorously shaken 15 times. Stools samples were collected and immediately transferred into a Omnigene-Gut tube, (DNA Genotek, 3000–500 Palladium Drive, Ottawa, ON, Canada, K2V 1C2) for stabilization of microbial DNA from feces for gut microbiome profiling. In the end, the sample was transferred jointly with the fecal specimen to the acceptance of the laboratory. Biological materials and extracted DNA were conserved at − 20 °C before any additional processing and kept at − 20 °C for a short period until library preparation and sequencing.
DNA extraction
DNA extraction from fecal samples was performed by resuspending into 1 ml of the ASL Stool lysis buffer (Qiagen GmbH, Germany) a tiny quantity of fecal material by vigorous pipetting. Instead, oral swab samples were resuspended in a 200 µl physiologic solution. Then the resuspended materials were processed to extract DNA using the MagDEA DNA 200GC extraction kit and PSS Magtration System 12GC automated platform, according to the manufacturer’s instructions (Precision System Science PSS Co., Ltd, Matsudo, Japan). DNA was eluted into 100 µl of 10 mM TE buffer; quality and quantity were evaluated by spectrophotometric and Qubit fluorimetric quantitation assays, respectively (ThermoFisher Scientific, Waltham, MA).
16S rRNA sequencing and data analysis
In detail, 3 ng of DNA was used for each 16S amplification reaction performed with Ion 16S Metagenomics Kit™ (ThermoFisher Scientific, Waltham, MA). It allows the analysis by PCR amplification of 7 out of 9 informative 16S polymorphic regions (V2, V4, V8, V3, V6-7, V9) according to the manufacturer protocols. IonPlus-Library kit for AB library builder (ThermoFisher Scientific, Waltham, MA) was used for library synthesis. Different bar-coded libraries were automatically handled and loaded onto an Ion 520 chip by the Ion Chef System and then sequenced by the GeneStudio S5 system (ThermoFisher Scientific, Waltham, MA).
Data analysis was performed with Ion Reporter ™ suite software (v 5.18) using both the curated Greengenes (v13.5) and the premium curated MicroSEQ ID 16S rRNA reference library (v2013.1) databases with standard parameters.
Bioinformatics and statistics
Compositional/functional profiling and the comparative analysis of microbiome data were performed with MicrobiomeAnalyst13,14,15. Data filtering for low abundance and low variance OTUs (based on the prevalence in 20% of samples and interquartile range or iqr set at 10%) was applied for all the relative abundance comparisons using different algorithms (metagenomeSeq, EdgeR, DESeq2, LDA, and LEfSe). All p-values have been adjusted to correct for multiple hypotheses, using Benjamini and Hochberg false discovery rate (FDR < 0.05), unless differently specified. In the comparison of cases and the controls, the SparCC (Sparse Correlations for Compositional Data) analysis was used16. SparCC is a computational tool that analyses the interactions between different microbial species in the microbiome. SparCC uses a technique called "sparse inverse covariance estimation" to identify patterns of interactions in the data and to infer the underlying network of interactions between the components. SparCC is often used to identify keystone species, which may participate in maintaining the overall eubiosis. MD-index (MDI) is the natural logarithm of the ratio of the sum of the abundance of all the taxa that are enriched, and the ones depleted17. The microbial dysbiosis index measures the imbalance or deviation of the microbial community from a healthy or normal state. The MDI is calculated using the relative abundance of different microbial taxa and comparing it to a reference or control population (in our case situation before transplant). The value of the number higher or lower than zero indicates a greater degree of dysbiosis or abnormal microbial composition, while a value around zero denoted a normal or healthy microbial population.
Ethical approval
Informed consent was obtained from all individual participant, or their family included in the study. The studies involving human participant were conducted in accordance with the Declaration of Helsinki and were reviewed and approved by the Ethics Committee of Regione Liguria 369/2018 issued for IRCCS Istituto Giannina Gaslini—Genova, PI Dr Maura Faraci.
Results
Patients
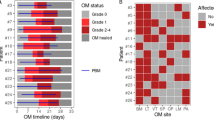
During the period between 2019 and 2022, 17 patients (median age 6.0 years, range 0.7–24.7 years) who underwent allogeneic HSCT were enrolled in the study, 9 of them affected by non-malignant diseases (3 bone marrow failures, 5 congenital immunodeficiencies, and one autoinflammatory disease) and 8 by malignant diseases (5 acute lymphoblastic leukemia and 3 acute myeloid leukemia) (Table 1). 8 patients received stem cells from a haploidentical donor, 5 from an alternative donor, and 4 from a related donor. A myeloablative (MAC) CR was used in 16 patients, including total body irradiation (TBI) in 5 patients, busulfan (Bus) in 3 patients, and treosulfan (Treo) in the remaining 8 patients. One patient received a reduced-intensity CR (RIC) for congenital dyskeratosis. Out of 17 patients enrolled in the study, 15 patients with age ≥ 2 years and with a primary or secondary dentition were included in the statistical analysis. In addition, the same cut-off was valid for estimating microbiome composition18.
In detail, 11 patients developed OM subdivided into grade 1 (n = 5), grade 2 (n = 3), grade 3 (n = 1), and grade 4 (n = 2). 7 patients who received HSCT for non-malignant developed mild OM (grade 0 in 4, grade 1 in 3, and grade 2 in 2), while patients transplanted for the malignant disease had more severe OM (grade 0 in 2, grade 1 in 2, grade 2 in 1, grade 3 in 1, grade 4 in 2). GI-mucositis was observed only in 2 patients (grade 2) who received HSCT for non-malignant disease. Acute GvHD (aGvHD) was observed in 8 patients, 4 of them had cutaneous acute GvHD above grade 2, and 3 with a GI aGvHD (grade 3). Limited chronic GvHD was present in one patient (Table 1).
Regarding nutrition, it is known that there are different recommendations for the use of enteral or parenteral feeding19. In our case, 15 patients received total parenteral nutrition for a median of 26.8 days (12–55 days).
Alpha and beta diversity indexes
The alpha diversity profiling indexes used to analyze the microbial community in oral samples were the Chao1, Shannon, or Simpson indexes. The first index provided evidence of a correlation among microbiome richness, while the Shannon and Simpson indexes provided information about community richness and evenness. Together, these metrics offer valuable insight into the structure and composition of the microbiome. All the indexes showed in the oral swabs the lowest values at the HSCT engraftment, while they increased gradually with the highest values after 100 days. All alpha indexes analyzed reached statistical significance in oral swab samples (Fig. 1a), but only the Chao1 reached significance in fecal samples (Fig. 1c).

Alpha diversity results comparing the complete cohort of oral (a) and fecal (c) microbiomes of patients > 2 years old. From the left, the alpha-indexes estimate the community richness (Chao1-index) or community richness and evenness (Shannon or Simpson indexes). The beta diversity Bray–Curtis’s distance matrix analysis for oral (b) and fecal (d) samples. The comparison performed with the Principal Coordinates Analysis (PCoA) showed using a 2D or 3D distribution. Permutational Multivariate Analysis of Variance (PERMANOVA) was used for statistical significance analysis.
According to the data, it appears that the samples taken before transplant from patients affected by malignant diseases, both in oral and stool, had lower values in all three alpha diversity indexes as compared to the samples from patients with non-malignant diseases. Based on the analysis performed at engraftment, it seems that there was a similar trend observed in the samples taken from patients affected by malignant diseases, with a single exception. The Chao1 index of oral microbiome resulted in higher values for samples from malignant diseases (seven patients, Chao1: 26.07 + / − 7.52 on) as compared to non-malignant diseases (seven patients, Chao1: 20.14 + / − 7.08).
Beta diversity was analyzed using the Bray–Curtis index and PERMANOVA statistical evaluation without reaching significance either in oral or fecal samples (Fig. 1b, d).
Oral and fecal microbiome analysis and taxonomic determination by 16S-targeted NGS sequencing
We analyzed 98 oral and fecal microbiomes that gave 194,521 ± 57,929 16S mapped reads per sample. These reads identified 1524 ± 551 operational taxonomic units (OTUs) in the 49 oral swab samples and 1545 ± 389 OTUs in the fecal samples analyzed. The analysis of oral microbiomes indicated that the abundance of the Firmicutes phylum increased over the time points studied (pre-HSCT, at engraftment, + 30 days, and + 100 days after HSCT), rising from 46 to 72%. However, the Proteobacteria and Bacteroidetes phyla showed the opposite trend, with a decrease in abundance from 20 to 10% and 17% to 9%, respectively. Accordingly, the analysis of Firmicutes/Bacteroidetes (F/B) or Firmicutes/Proteobacteria (F/P) ratios showed that both increased their value along the four observations, with F/B ranging from 2.7 to 8.0 and F/P from 2.3 to 7.2 (Table 2a). The abundance of Fusobacteria halves after 100 days from transplant only, according to Table 2a. On the other hand, the fecal microbiomes showed a different behavior, with Firmicutes maintaining a stable abundance rate, Bacteroidetes increasing, and Proteobacteria decreasing their fullness along longitudinal points, as shown in Table 2b.
The findings of the analysis of taxa abundances in oral or fecal microbiomes at engraftment compared to the situation before HSCT suggest that there were significant changes in the abundance of some genera in both oral and fecal samples (Fig. 2a and b, Supplementary material Table S1). For instance, the genera Abiotrophia and Streptococcus were more significantly abundant at engraftment in both oral and fecal samples. It’s interesting to note that some genera such as Atopobium, Rothia, Actinomyces, Gemella, Granulicatella, and Escherichia were significantly more abundant in the fecal microbiome at engraftment, while the Corynebacterium, Serratia, Campylobacter, Catonella, Peptostreptococcus, Veillonella, and Porphyromonas were found to be more abundant at engraftment in oral microbiomes. Additionally, the findings showed that Prevotella, Dorea, Pseudoflavonifractor, Bifidobacterium, Blautia, Faecalibacterium, Eubacterium, and Roseburia were significantly more abundant before HSCT in fecal samples only, and the same for Tannerella, and Stomatobaculum was in oral microbiomes. After 30 days following transplant, the genera Gemella and Streptococcus were found to be significantly more abundant in both oral and fecal samples as shown in Fig. 2c and d, respectively (Supplementary material Table S1). At 30 days after HSCT, Abiotrophia and Rothia were the genera with the highest value of abundance increase in fecal microbiomes. On the other hand, in oral microbiomes, Veillonella and Serratia were found to be more abundant at + 30 days. Finally, the genera typically more abundant in samples before HSCT were Prevotella and Dorea in fecal microbiome comparisons (Fig. 2).

Longitudinal analysis using the EdgeR method for differential expression analyses was conducted to identify the difference in abundance of various taxa between oral (a, c, e) and stool (b, d, f) samples at different time points always compared with the microbiome of samples before HSCT transplant. All the analyses were focused on the genus level. The numbers in squared parenthesis represent the number of patients belonging to the indicated group of samples. The fold change value is represented logarithmically on base 2 (log2FC), and it shows the increase or decrease of abundance of a particular taxa in the comparison between the two groups of samples. FDR (False Discovery Rate) indicates the statistical significance value after adjustment for multiple comparisons. All statistical analyses showed FDR < 0.05, * < 0.01, or ** < 0.001.
Microbiome associated with oral mucositis
Oral mucositis (OM) is a common complication associated with hematopoietic stem cell transplantation (HSCT). OM refers to the inflammation and ulceration of the mucous membranes lining the mouth, throat, and gastrointestinal tract. Occurs because of the high-dose chemotherapy and/or radiation therapy that patients undergo as part of the transplant conditioning regimen. The severity of oral mucositis can vary, ranging from mild discomfort to severe pain, and it can significantly impact a patient’s quality of life. It’s important to note that both malignant and non-malignant disease patients experienced complications such as OM. This emphasizes the importance of implementing personalized care and treatment plans that are specifically designed to address the unique health needs of each patient. Furthermore, the analysis of phyla-abundances during engraftment revealed a strong positive correlation between the increasing expansion of Fusobacteria and the severity of oral mucositis, highlighting the need for further research into the underlying mechanisms driving this relationship. It’s interesting to note that patients who did not exhibit OM had only 2% of Fusobacteria in their samples (Fig. 3). According to the data, the abundance of Fusobacteria increased from 8 to 18% in OM-positive specimens, with the highest percentage found in more severe cases of mucositis (grade ≥ 3), as shown in Fig. 3. It’s interesting to note that a longitudinal analysis of differential abundances in oral samples with mucositis (all grades) showed that Mycoplasmatales descendants and the Prevotella oris species were among the taxa with the most statistically significant increase in specimens at engraftment or after 30 days from HSCT, respectively. (Fig. 4a, Supplementary material Table S4). Moreover, in the oral microbiome, Mycoplasma (at engraftment) and Prevotella oris (after 30 days from transplant) were also associated with the best statistical correlation with grade ≥ 2 OM in oral swab samples (Fig. 4b, Supplementary material Table S5). On the contrary, Ruminococcaceae, Rothia, and the Streptococcus genus in oral swab samples were found relevant, from a statistical point of view, in patients that never reported oral mucositis, so to be considered as protective taxa (Figs. 4a, 3b, Supplementary material Tables S4, and S5).

Phila abundances in oral swabs at engraftment in patients treated with myeloablative conditioning (MAC). Microbiome from Oral-mucositis (OM) negative samples (a) compared to OM positive (all grades) specimens (b), OM positive grades > = 2 (c), or affected by severe mucositis (OM grades > = 3) in panel. (d) In square brackets, it was indicated the number of patients belonging to the groups in the analysis. A horizontal line splitting each box in two indicates the median. The box limit indicates the upper and lower quartiles of the data.

(a) The figure showed the DESeq2 analysis to identify the difference in abundance of various taxa between oral samples that developed oral mucositis of all grades, either at engraftment (10 patients) or (b) after 30 days from it (7 patients) and specimens that never had mucositis (5 patients). The fold change value, represented as logarithmic on base 2 (log2FC), shows the increase or decrease of abundance of a particular taxa in the comparison between the two groups of samples. FDR (False Discovery Rate) indicates the statistical significance value after adjustment for multiple comparisons. All statistical analyses showed FDR < 0.001, except for the taxa indicated with an asterisk (*) that had FDR < 0.05.
On the contrary, the analysis of fecal microbiome showed Egghertella lenta, Bacteroides ovatus, Ruminococcus gnavus, Lachnoclostridium lavalense, Pseudoflavonifractor, and different species of Clostridium always associated with specimens of reporting oral mucositis (Fig. 4, Supplementary material Tables S4, and S5).
Networks of interaction among microbial taxa in oral mucositis
The network correlation between the taxa abundance and OM was also studied in the oral microbiome using sparse correlation for compositional data (SparCC) analysis. The MD-Index for each informative comparison was also calculated. Thus, in patients undergoing the myeloablative (MAC) regimen, the MD-Index at engraftment of cases of patients developing mucositis of any complexity, compared to samples from patients that never developed mucositis analyzed at the level of genera was − 3.1939 (Fig. 5a) and − 1.5818, the one associated with mucositis with more complexity (grades ≥ 2). These data indicated that an increased MD-Index value correlates with more complex mucositis. The SparCC analysis of the microbiome associated with OM of any grade indicated that 29 genera produce positive/negative correlations (threshold 0.3, p ≤ 0.05). Among these genera, two third (at engraftment) and approximately three-quarters of 30 genera (after 30 days from HSCT) showed positive correlations among them. Overall, the Prevotella genus showed the highest number of correlations with other taxa (seven), computed by the SparCC network analysis (Fig. 5a). The Capnocytophaga showed six correlations all only with positive sign. Interestingly, the species Atopobium parvulum and Capnocytophaga sputigena confirmed their association with mucositis (Fig. 5b), as already suggested in patients treated with radiotherapy for haematologic malignancies20,21. More, the analysis of the microbial species associated with OM (grades ≥ 2) in patients subjected to MAC regimen showed a network of 178 species (threshold of 0.3 and a p ≤ 0.05), 106 of them producing positive correlations and the MD-Index of cases over controls after 30 days from transplant was 0.6979 (Fig. 5b). Streptococcus, Prevotella, and Veillonella were among the genera showing the highest correlation numbers with other taxa in the network analysis after 30 days from transplant. In addition, 12, 10, and 8 were the species found to belong to these genera. Thus, indicating their importance in the association with OM. Overall, these network analyses on oral microbiome pinpointed that Prevotella correlates positively with OM samples, while Streptococcus was always associated with a negative correlation.

(a) SparCC analysis of oral microbiome at engraftment comparing samples with mucositis (of any grade) to ones negative for such complication. All patients underwent a myeloablative conditioning regimens (MAC). Each node represents genera that were differently colored in consideration of the preferential abundance in mucositis-positive (orange) or mucositis-negative samples (green). Red lines connecting nodes indicate a positive correlation between taxa, while blue lines showed a negative one. MD-index represents the dysbiosis-index of cases over controls. Network was computed for p < = 0.05 and correlation threshold was set at 0.3. (b) Oral microbiome after 30 days from transplant among samples with severe oral mucositis (grades > = 2) compared to samples negative for such complication. Each node represents a species differently colored in consideration of the preferential abundance in mucositis positive (orange) or mucositis negative samples (green). Red lines connecting nodes indicate a positive correlation between taxa, while blue lines show a negative one. MD-index represents the dysbiosis-index of cases over controls. Network was computed for p < = 0.05 and the correlation threshold was set at 0.5.
Acute GVHD
Regarding aGvHD, the longitudinal analysis of Chao1, Shannon, and Simpson’s alpha diversity indexes showed higher median values on aGvHD samples (Fig. 6). Simpson index was the only measure to reach statistical significance, at least in the pre-HSCT specimens (p = 0.0350) (Fig. 6). At engraftment, the compositional microbiome analysis showed a sturdy increase in the relative abundance of Proteobacteria in patients with aGvHD from 17 to 49% (Fig. 7). More in detail, the Firmicutes/Proteobacteria ratio was 3.2 in samples from patients without cutaneous aGvHD and 0.5 in cutaneous aGvHD patients, thus indicating an unbalanced increase of Proteobacteria compared to the Firmicutes in aGvHD patients. Differently from the patients who developed mucositis, the fecal microbiome of subjects with cutaneous aGvHD was associated with the Proteobacteria phylum (Fig. 8, Table S6). This data might indicate either a direct dependency of the showed microbial family with aGvHD or an association with pro-inflammatory taxa, as suggested by others22. More in detail, different genera and species showed a positive correlation with cutaneous aGvHD in stool samples, like some species of Enterobacteriaceae like Klebsiella, Kluyvera, Yersinia, Serratia, and Enterobacter, as it was also confirmed by the network correlation analysis performed with SPARCC algorithms (Fig. 9). At 30 days after HSCT, among the bacterial taxonomy with higher abundance in stool samples from patients suffering from cutaneous aGvHD, different of them belong to the Alphaproteobacteria (Gemminger formicilis) and Deltaproteobacteria (Bilophila wadsworthia) (Fig. 8 and Supplementary material Table S6), followed by the ones belonging to the Betaproteobacteria (Parasutterella), and Bacteroides genus (Fig. 8 and Supplementary material Table S6). Indeed, the Firmicutes/Bacteroidetes ratio always showed a lower value in patients with cutaneous aGvHD compared to the ones that did not develop it at all the time points studied (Fig. 7).

The results show a comparison of the fecal microbiome in patients over 2 years old. The comparison is between the microbiome of samples that underwent cutaneous aGvHD and those that never developed such complications. The indexes in the graph estimate community richness (Chao1-index) or community richness and evenness (Shannon or Simpson indexes) from left to right. (a) Data before HSCT; (b) samples at engraftment; and (c) samples at 30 days after transplant.

Phila abundances in fecal microbiome samples at engraftment, comparing patients that suffered from acute Graft Versus Host Disease (aGvHD) shown on the right with others that never showed such complications (on the left). In square brackets, it was indicated the number of patients belonging to the groups compared in the analysis. A horizontal line splitting each box in two indicates the median. The box limit indicates the upper and lower quartiles of the data.

The figure showed the DESeq2 analysis to identify the difference in abundance of various taxa between oral samples that later developed acute GvHD (aGvHD), either before HSCT (6 patients) at engraftment (5 patients) or after 30 days from it (4 patients), and specimens that never had mucositis (7, 7 and 5 patients, respectively). The fold change value, represented as logarithmic on base 2 (log2FC), shows the increase or decrease of abundance of a particular taxa in the comparison between the two groups of samples. FDR (False Discovery Rate) indicates the statistical significance value after adjustment for multiple comparisons. All statistical analyses showed FDR < 0.001, except for the taxa indicated with ^, FDR < 0.01, or with an asterisk (*) that had FDR < 0.05.

Fecal microbiome from patients treated with myeloablative regimen at HSCT engraftment. Acute Graft Versus Host Disease (aGvHD) positive samples were compared to negative ones for such complications. Each node represents genera that were differently colored in consideration of the preferential abundance in aGvHD samples (orange) or aGvHD-negative samples (green). Red lines connecting nodes indicate a positive correlation between taxa, while blue lines showed a negative one. MD-index represents the dysbiosis-index of positive cases over negative ones. Network was computed for p < = 0.05 and correlation threshold was set at 0.3.
Discussion
This study aims to investigate the changes in the microbiome of the oral and intestinal microbiomes in pediatric patients who underwent allogeneic HSCT. The study found that the toxic effects of different conditioning regimens used during HSCT can lead to microbial dysbiosis in various environments, including the oral cavity23. This can result in the development of infectious and immunological complications in patients. The study highlights the importance of monitoring the microbiome in HSCT patients, especially in the oral cavity, to prevent complications and ensure successful outcomes. The peculiarity of this study is the detailed analysis of the oral microbiome after HSCT, which is rarely reported in the literature24. Compositional analysis of the oral bacterial phyla demonstrated a heavy expansion of Fusobacteria in patients with OM. Indeed, samples from patients affected by severe mucositis (grade ≥ 3) showed an abundance of the genus Fusobacterium of 18%, while the same taxa displayed only 2% in patients that never developed OM. All three alpha diversity profiling indexes (Chao1, Shannon, and Simpson) suggested the susceptibility of both oral and fecal microbiomes to the chemo and/or radiotherapy administered during the CR. Indeed, the used indexes had the lowest value at engraftment and reached the highest score 100 days after transplant, as a flourish of microbiota over time. Our data show that a decrease in taxa richness and evenness was associated with CR for oral microbiome analysis, whereas the fecal specimens showed a statistically relevant decrease in richness (Chao-1) only. Longitudinal analysis of the oral and fecal microbiomes showed a mutually exclusive presence of taxa in one of the two studied environments, with few exceptions. In patients affected by oral mucositis, the oral microbiome showed an increased abundance of different species of the Fusobacterium, Mycoplasma, Capnocytophaga, and Prevotella and a depletion of Streptococcus, Granulicatella, and Rothia taxa. This is in line with the findings of previous reports2,20. In contrast, the gut microbiome showed enrichment of Eggerthella lenta and depletion of Streptococcus, Rothia, Actinomyces, Granulicatella, and Veillonella species in subjects presenting mucositis (grade ≥ 2). Our data mainly agree with previous reports2,25. Moreover, our data on differential abundance and the SparCC network analysis showed an association between the Atopobium genus and its species Atopobium parvulum with mucositis of grade ≤ 2.
It has been observed that the Atopobium parvulum species has been reported by others to be positively associated with mucositis incidence26. It is interesting to note that our data on oral microbiomes indicates that deciphering microbial interactions through network analysis could help identify key bacteria for both health and disease. However, it’s worth mentioning that our findings contradict, at least in part, those of other authors26, who have also suggested that the genus Kingella is associated with mucositis. In our study, Kingella was mainly found in samples from patients who had not experienced oral mucositis. This suggests that further research is needed to fully understand the role of Kingella in oral health and disease.
Our data showed that Alphaproteobacteria (Gemminger), and Deltaproteobacteria (Bilophila) were positively associated with aGvHD. The role of Escherichia, a member of Gammaproteobacteria, in supporting cutaneous aGvHD is quite contradictory since our data mainly found a negative correlation with cutaneous aGvHD, differently from what has been already reported by other groups27,28. Our data could be explained by the effect of some metabolites secreted by Escherichia coli, like indole and its derivatives, that were known to mitigate GvHD29. Also, Prevotella descendants and Bacteroides were negatively associated with aGvHD (Supplementary material Table S6), as suggested by a previous report22. However, in an animal model, others found a positive correlation between Prevotella ssp. and aGvHD28. In addition, the behavior of the Clostridiales was the opposite of what was already presented10, except for the Anaerostipes genus, which in our data showed a negative correlation with cutaneous aGvHD.
Our research, as well as studies conducted by others, indicate that the imbalance of microbes caused by CR (conditioning regimen) can result in complications during hematopoietic stem cell transplantation (HSCT), such as mucositis and cutaneous acute graft-versus-host disease (aGvHD). To prevent these conditions, several methods have been tested, including the use of probiotics, pre/postbiotics, nutritionally active substances, and various species of lactic acid bacteria30,31,32,33. These approaches have shown promise in reducing the incidence of CR-induced oral mucositis33. Additionally, other studies have identified bacterial taxa associated with the development of mucositis and its severity and the role of dysbiosis during preconditioning4,34. It may be possible that studies in this field are paving the way toward more personalized and effective treatments by evaluating bacterial signature profiles might lead to avoiding or decreasing the severity of HSCT complications.
Data availability
The datasets presented in this study are openly available in the Short Read Archive (SRA), Sequence Read Archive NCBI-NIH. SRA_BioProject ID PRJNA929702.
Abbreviations
- HSCT:
-
Hematopoietic stem cell transplantation
- OM:
-
Oral mucositis
- aGvHD:
-
Acute graft versus host disease
- GvHD:
-
Graft versus host disease
- CR:
-
Conditioning regimen
- GI:
-
Gastrointestinal
- TBI:
-
Total body irradiation
- Treo:
-
Treosulfan
- FDR:
-
False discovery rate
- SparCC:
-
Sparse correlations for compositional data
- MD-index:
-
Microbial dysbiosis-index
- MAC:
-
Myeloablative regimen
- RIC:
-
Reduced intensity conditioning
- OTUs:
-
Operational taxonomic units
- F/B:
-
Firmicutes/Bacteroidetes
- F/P:
-
Firmicutes/ProteobacteriA
- SRA:
-
Short read archive
- PCoA:
-
Principal coordinates analysis
References
Pignatelli, P. et al. Microbiota and oral cancer as a complex and dynamic microenvironment: A narrative review from etiology to prognosis. Int. J. Mol. Sci. 23, 8323. https://doi.org/10.3390/ijms23158323 (2022).
Hong, B. Y. et al. Chemotherapy-induced oral mucositis is associated with detrimental bacterial dysbiosis. Microbiome 7, 66. https://doi.org/10.1186/s40168-019-0679-5 (2019).
Sonis, S. T. The pathobiology of mucositis. Nat. Rev. Cancer 4, 277–284. https://doi.org/10.1038/nrc1318 (2004).
Bruno, J. S. et al. Commensal oral microbiota impacts ulcerative oral mucositis clinical course in allogeneic stem cell transplant recipients. Sci. Rep. 12, 17527. https://doi.org/10.1038/s41598-022-21775-3 (2022).
Heidrich, V. et al. Dental biofilm microbiota dysbiosis is associated with the risk of acute graft-versus-Host disease after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 2021(12), 692225. https://doi.org/10.3389/fimmu.2021.692225 (2021).
Elad, S. et al. MASCC/ISOO clinical practice guidelines for the management of mucositis secondary to cancer therapy. Cancer 126, 4423–4431. https://doi.org/10.1002/cncr.33100 (2020).
Al-Qadami, G., Van Sebille, Y., Bowen, J. & Wardill, H. Oral-gut microbiome axis in the pathogenesis of cancer treatment-induced oral mucositis. Front. Oral Health 3, 881949. https://doi.org/10.3389/froh.2022.881949 (2022).
Chaudhry, H. M. et al. The incidence and severity of oral mucositis among allogeneic hematopoietic stem cell transplantation patients: A systematic review. Biol. Blood Marrow Transpl. 22, 605–616. https://doi.org/10.1016/j.bbmt.2015.09.014 (2016).
Peterson, D. E., Srivastava, R. & Lalla, R. V. Oral mucosal injury in oncology patients: perspectives on maturation of a field. Oral Dis. 21, 133–141. https://doi.org/10.1111/odi.12167 (2015).
Lin, D. et al. Roles of the intestinal microbiota and microbial metabolites in acute GVHD. Exp. Hematol. Oncol. 10, 49. https://doi.org/10.1186/s40164-021-00240-3 (2021).
Przepiorka, D. et al. 1994 Consensus conference on acute GVHD grading. Bone Marrow Transpl. 15, 825–828 (1995).
Filipovich, A. H. et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol. Blood Marrow Transpl. 11, 945–956. https://doi.org/10.1016/j.bbmt.2005.09.004 (2005).
Dhariwal, A. et al. MicrobiomeAnalyst—A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180-188. https://doi.org/10.1093/nar/gkx295 (2017).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821. https://doi.org/10.1038/s41596-019-0264-1 (2020).
Lu, Y. et al. MicrobiomeAnalyst 2.0: Comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 51, W310–W318. https://doi.org/10.1093/nar/gkad407 (2023).
Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687. https://doi.org/10.1371/journal.pcbi.1002687 (2012).
Wei, S., Bahl, M. I., Baunwall, S. M. D., Hvas, C. L. & Licht, T. R. Determining gut microbial dysbiosis: A review of applied indexes for assessment of intestinal microbiota imbalances. Appl. Environ. Microbiol. 87, e00395-e421. https://doi.org/10.1128/AEM.00395-21 (2021).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–8. https://doi.org/10.1038/s41586-018-0617-x (2018).
D’Amico, F. et al. Enteral nutrition in pediatric patients undergoing hematopoietic SCT promotes the recovery of gut microbiome homeostasis. Nutrients 11, 2958. https://doi.org/10.3390/nu11122958 (2019).
Vesty, A. et al. Oral microbial influences on oral mucositis during radiotherapy treatment of head and neck cancer. Support Care Cancer 28, 2683–2691. https://doi.org/10.1007/s00520-019-05084-6 (2020).
Funada, H. et al. Capnocytophaga sputigena bacteremia associated with acute leukemia. Kansenshogaku Zasshi 67, 622–628. https://doi.org/10.11150/kansenshogakuzasshi1970.67.622 (1993).
Taur, Y. et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182. https://doi.org/10.1182/blood-2014-02-554725 (2014).
Heidrich, V. et al. Longitudinal analysis at three oral sites links oral microbiota to clinical outcomes in allogeneic hematopoietic stem-cell transplant. Microbiol. Spectr. 11, e0291023. https://doi.org/10.1128/spectrum.02910-23 (2023).
de Molla, V. C. et al. Disruption of the oral microbiota is associated with a higher risk of relapse after allogeneic hematopoietic stem cell transplantation. Sci. Rep. 11, 17552. https://doi.org/10.1038/s41598-021-96939-8 (2021).
Schincaglia, G. P. et al. Clinical, immune, and microbiome traits of gingivitis and peri-implant mucositis. J. Dent. Res. 96, 47–55. https://doi.org/10.1177/0022034516668847 (2017).
Shouval, R. et al. Patterns of salivary microbiota injury and oral mucositis in recipients of allogeneic hematopoietic stem cell transplantation. Blood Adv. 4, 2912–2917. https://doi.org/10.1182/bloodadvances.2020001827 (2020).
Eriguchi, Y. et al. Graft-versus-host disease disrupts intestinal microbial ecology by inhibiting Paneth cell production of α-defensins. Blood. 120, 223–231. https://doi.org/10.1182/blood-2011-12-401166 (2012).
Heimesaat, M. M. et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut 59, 1079–1087. https://doi.org/10.1136/gut.2009.197434 (2010).
Swimm, A. et al. Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease. Blood 132, 2506–2519. https://doi.org/10.1182/blood-2018-03-838193 (2018).
Hassan, H., Rompola, M., Glaser, A. W., Kinsey, S. E. & Phillips, R. S. Systematic review and meta-analysis investigating the efficacy and safety of probiotics in people with cancer. Support Care Cancer 26, 2503–2509. https://doi.org/10.1007/s00520-018-4216-z (2018).
Suez, J., Zmora, N., Segal, E. & Elinav, E. The pros, cons, and many unknowns of probiotics. Nat. Med. 25, 716–729. https://doi.org/10.1038/s41591-019-0439-x (2019).
Feng, J. et al. Oral administration of probiotics reduces chemotherapy-induced diarrhea and oral mucositis: A systematic review and meta-analysis. Front. Nutr. 9, 823288. https://doi.org/10.3389/fnut.2022.823288 (2022).
Caluwaerts, S. et al. AG013, a mouth rinse formulation of Lactococcus lactis secreting human Trefoil Factor 1, provides a safe and efficacious therapeutic tool for treating oral mucositis. Oral Oncol. 46, 564–570. https://doi.org/10.1016/j.oraloncology.2010.04.008 (2010).
Laheij, A. M. G. A. et al. Long-term analysis of resilience of the oral microbiome in allogeneic stem cell transplant recipients. Microorganisms 10, 734. https://doi.org/10.3390/microorganisms10040734 (2022).
Acknowledgements
We would like to thank Umberto Buelli and Gianni Berretta, SIA—IstitutoGianninaGaslini for helping in the computer network support.
Funding
Funding support was provided by Università degli studi di Genova: Prof.ssa F. Angiero, Dott. A. Trevisiol, Dott. L. Rufino, Università degli studi di Milano: Prof. S. Blasi, Istituto Giannina Gaslini: Dr. Roberto Biassoni Ricerca Corrente Grant from the Ministry of Health and from Compagnia di San Paolo—2019.0880—ID ROL 32612.
Author information
Authors and Affiliations
Contributions
Each author participate and contribute at different level to the conception or design of the work; or the acquisition, analysis, or interpretation of data, drafted the work or substantively revised it; AND has approved the submitted version (and version substantially edited by journal staff that involves the author’s contribution to the study); AND agrees to be personally accountable for the author’s own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and documented in the literature. Conceptualization—R.B., M.F., S.B., F.A., A.T., and L.R. Methodology—R.B., C.B., E.D.M., E.O., M.F. Software—R.B., M.S.; Validation—R.B., C.B., E.D.M., and M.F. Formal Analysis—R.B., C.B., M.S. Investigation—M.F., F.P., G.D., S.G., A.T., L.R. Resources—R.B., C.B., E.D.M.; Data Curation—R.B., M.S.; Writing—Original Draft Preparation—R.B., M.F. Writing, Review and Editing—R.B., M.F., F.P., G.D., S.G., S.B., A.T., L.R. Visualization, R.B.; Supervision, R.B., M.F.; Project Administration, M.F., R.B. Funding Acquisition, F.A., G.F., R.B., M.F.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Faraci, M., Bonaretti, C., Dell’Orso, G. et al. Association between oral and fecal microbiome dysbiosis and treatment complications in pediatric patients undergoing allogeneic hematopoietic stem cell transplantation. Sci Rep 14, 6708 (2024). https://doi.org/10.1038/s41598-024-55690-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55690-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
