
Abstract
The marine littoral earthworm Pontodrilus litoralis (Grube, 1855) is widely distributed and is reported as a single species. This study utilized an integrative taxonomic approach based upon morphological examination, phylogenetic reconstruction, and molecular species delimitation, to test whether the taxon is a single species or a species complex. For this, a total of 114 P. litoralis specimens collected from North America, Africa, Australia and Oceania, Europe and Asia were used. The phylogenetic analyses revealed deeply divergent mitochondrial lineages and a high level of genetic diversity among P. litoralis populations. Both single and multi-locus species delimitation analyses yielded several molecular operational taxonomic units. Therefore, due to the homogeneity of morphological characteristics, it is likely that the morphospecies P. litoralis is a complex of four or more cryptic species, suggesting that more sampling is required and that the population structure genetic data and gene flow need to be investigated.
Similar content being viewed by others

Introduction
The bioluminescent earthworm Pontodrilus litoralis (Grube, 1855) has been reported as a cosmopolitan species, inhabiting marine littoral ecosystems in the sub-temperate and tropical coastal areas of the Atlantic, Pacific, and Indian oceans1,2,3,4, and is reported to be both arenicolous and limicolous. The first description of this littoral earthworm, named as Lumbricus litoralis by Grube (1855)5, was based on the morphological characteristics of a Mediterranean sample from the Villefranche-sur-Mer (formerly Villafranca) on Côte d’Azur, France. The genus Pontodrilus was first established by Perrier (1874)6 who also described P. marionis Perrier, 1874; however, Beddard (1895)7 subsequently synonymized P. marionis with L. litoralis. Easton (1984)1 then provided an extensive list of P. litoralis synonyms and references to the taxonomic literature and concluded that P. litoralis is a single species which is highly variable. Although, a few other morphologically distinct species of Pontodrilus have been discovered, only two species of Pontodrilus, including P. litoralis, have been reported from Thailand and peninsular Malaysia2. Chen et al. (2021)8 hypothesized that the widespread populations of P. litoralis throughout the world resulted from their transport by currents, which is congruent with Blakemore’s (2007)9 suggestion that the wide distribution of P. litoralis is due to the transport of ships’ sand-ballast, and the natural rafting of euryhaline cocoons. The wide range of salinity tolerance of P. litoralis, shown experimentally by Seesamut et al., (2022)10, may have facilitated this species’ wide distribution pattern.
Molecular (DNA) taxonomy in earthworms has mostly used a single marker gene, in particular the mitochondrial cytochrome c oxidase subunit 1 (COI) gene. When such a marker is used to identify species, the method is referred to as DNA barcoding11,12,13,14,15. However, many earthworm studies have implemented both nuclear and mitochondrial genes in phylogenetic species delimitation16,17,18,19. Widely used methods based on single-locus sequences are, e.g., Automatic Barcode Gap Discovery (ABGD)20, Assemble Species by Automatic Partitioning (ASAP)21, Bayesian implementation of Poisson Tree Processes model (bPTP)22 and General Mixed Yule Coalescent model (GMYC)23; for more details, see the review by Martinsson & Erséus (2021)24 and Goulpeau et al., (2022)25. However, for sexually reproducing species, multiple-locus delimitation, which takes the evolution of more than one gene into account, may be more reliable for testing hypotheses of speciation events; for instance, congruent nodes in the comparison between one nuclear and one mitochondrial gene tree are more supportive of a speciation event (ceased gene flow) than are incongruent nodes, which are evidence of gene flow between individuals belonging to different “mitochondrial” (= maternal) lineages19,26,27.
Despite the worldwide distribution records of P. litoralis, scientists still believe that it is a single species, and this is largely based on morphological characteristics. Variation in the body size between populations in Asia has been studied, but these marked difference in the morphometrics of P. litoralis across geographic populations did not correlate with their genetic differences (COI). Rather, it was suggested that P. litoralis is a single species3. In this study, we aimed to test the hypothesis that the worldwide distributed earthworm P. litoralis is a single species as proposed by Easton (1984) and Seesamut (2019)1,3. The earthworms were collected from North America, Australia and Oceania, Europe, Africa, and Asia (East and Southeast Asia), and morphological examination, phylogenetic analysis, and species delimitation using the methods mentioned above plus multi-locus delineation using Bayesian phylogenetics and phylogeography28,29 were conducted.
Results
We obtained a total of 114 COI sequences of P. litoralis which included 22 specimens from North America, three from Africa, 12 from Australia and Oceania, three from Europe, and 74 from Asia (24 from East Asia and 50 from Southeast Asia) (Fig. 1, Table 1). The final aligned dataset, comprised of 658 bp sequence fragments, contained a total of 392 invariable (monomorphic) sites, 210 variable (polymorphic) sites (total number of mutations is 283), and 119 parsimony informative sites. The result yielded a total of 52 haplotypes, with a haplotype (gene) diversity of 0.978 and a nucleotide diversity (Pi) of 0.09838. All sequences are deposited in GenBank (Table 1). Based on P. litoralis samples from different geographic distributions, the COI phylogenetic tree revealed a high genetic diversity, and the COI-based species delimitations revealed that the 114 specimens were divided into 19 MOTUs by ABGD and ASAP, whereas the bPTP and GMYC methods yielded 30 and 31 MOTUs, respectively (Fig. 2).

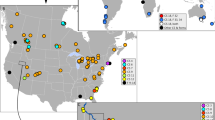
(A and B) Map showing the sampling sites of P. litoralis. The map is based on a map from D-maps (available at https://d-maps.com/carte.php?num_car=3228&lang=en), map was edited in Adobe Photoshop. (C) Photograph of P. litoralis from Thailand (photograph by Teerapong Seesamut).

A ML phylogenetic tree of P. litoralis based on the COI fragment sequence (658 bp) and the species delimitation clustering results. The nodes with ML bootstraps > 70% are considered well-supported. The scale bar indicates the branch length. ABGD, automated barcode gap discovery; ASAP, Assemble Species by Automatic Partitioning; bPTP, Bayesian implementation of Poisson Tree Processes model; GMYC, generalized mixed Yule coalescent model. The numbering is the input MOTUs of the BPP analyses, the letters a—d are the four most conservative MOTUs suggested by BPP.
The COI marker showed a higher variability than the ITS2. The COI haplotype network shows that 52 haplotypes were detected in 114 individuals, with each (location) population having its own single haplotype. Only one haplotype was shared across two locations from different countries: Quangbinh (Vietnam) and Taiwan (Fig. 3A). The ITS2 haplotype network showed a total of 36 haplotypes from 98 individuals (Fig. 3B). The highest numbers of mutational steps are 77 and 16 in COI and ITS2, respectively.

Haplotype networks for (A) COI sequences (658 bp) and (B) ITS2 sequences (437 bp) of P. litoralis. Lines with dashes and numbers between circles represent the number of mutational steps between two haplotypes. The number of samples in each haplotype corresponds to the size of the circles in the legend.
The phylogenetic relationships observed in the analysis of the concatenated data (COI + ITS2) were congruent with the COI and ITS2 phylogenetic trees (Figs. 2, 4, and Supplementary Fig. 1). The results of the BPP analyses are summarised in Table 2. In analysis A, B, and C, 17, 3, and 11, respectively, out of the 30 MOTUs are supported with a PP of > 0.95. The only two MOTUs that are supported in all three analyses are MOTU 29 and 30. In one of the three separate analyses of B and C, respectively, maximum support was found for combining a majority of the MOTUs into one. The most conservative estimate would be four MOTUs, i.e., (a) combining MOTUs 1–25, (b) combining MOTUs 26–28, (c) MOTU 29, and (d) MOTU 30 (Figs. 2, 4). There is some support for combining (i) MOTUs 26 and 27 and (ii) MOTUs 26 and 28. Based on these four MOTUs delineated by the BPP, interspecific COI uncorrected p-distances were calculated, revealing that the genetic divergence among this conservative set of MOTUs ranged from 13.9 to 16.9%.

A ML concatenated tree of COI and ITS2. Nodes with ML bootstraps > 70% are considered well-supported. The scale bar indicates the branch length. The four most conservative MOTUs suggested by the BPP analysis are marked with black circles labelled with a–d respectively.
Most P. litoralis specimens in this study were in the adult stage, and specimens from different collecting localities showed no difference in any distinctive morphological characteristics.
Discussion
Morphological investigation showed that the external and internal morphology of P. litoralis samples in this study correspond to the original description and those recently reported1,2,5,30. The analyses of the single-locus phylogeny and mitochondrial species delimitation suggested that P. litoralis is a complex of species, which all seem to be cryptic because of the homogeneity in their morphological characteristics. Moreover, a high degree of genetic structuring among different geographical populations of P. litoralis is evident. The occurrence of cryptic species in clitellates has frequently been uncovered, which is not surprising as there are few diagnostic morphological features that can be used to distinguish different species31. On the other hand, Martinsson et al. (2020)32 tested the species hypotheses of the enchytraeid worm Fridericia magna in Norway and Sweden and concluded that the data for this morphospecies is consistent with it being a single species. This and other examples (below) have shown that high intraspecific mitochondrial genetic distances are also common in clitellates.
In the semi-aquatic freshwater earthworm genus Glyphidrilus, ten single and multi-locus species delimitation methods revealed a high degree of incongruence between the genetic structures and morphology-based species identifications19. Several publications have examined and reported deeply divergent mitochondrial lineages and a high genetic diversity within well-established earthworm morphospecies14,17,33,34,35. Although the COI species delimitation analyses in this study suggested the presence of either 19, 30, or 31 MOTUs within P. litoralis, Lohse (2009)36 mentioned that geographic population structure is likely to lead to the overestimation of species numbers retrieved from species delimitation analyses. This has also been a critique of the multispecies coalescent methods, such as BPP37, and it is possible that this is a reason for our BPP analysis supporting about 20 MOTUs in the majority of runs, but then shifting to supporting much fewer MOTUs in some of the runs. This variation makes the interpretation of the results harder, and we have, therefore, chosen the more conservative estimate of MOTUs. Thus, we suggest that several MOTUs of P. litoralis are possibly affected by the bias from those species delimitation methods that analyzed the dataset containing different geographic populations of P. litoralis. With respect to the widespread distribution of the littoral earthworm P. litoralis, it may be dispersed around the world by humans or naturally be transported by currents8,9. Here, we suggest that the cosmopolitan distribution of P. litoralis is more likely to be caused by currents as human-mediated dispersal might cause the identical haplotype to be shared across different populations from distant locations38, while in our case there is a lack of identical haplotype shared across distant locations (Fig. 3).
For earthworms, we agree that 13% or thereabouts of COI interspecific genetic distance between two earthworm MOTUs could be used as a rule-of-thumb threshold to delimit different species14,19. Therefore, the most conservative recognition of only four MOTUs retrieved from the BPP analysis would suggest that P. litoralis is represented by four different species in our study (lineages a–d in Figs. 2 and 4). However, a much higher number of MOTUs of P. litoralis were detected by the different species delimitation methods. There are more than 20 synonyms of P. litoralis that have been reported from around the world1,2. Thus, in order to assign which synonym belongs to which different clade within the P. litoralis species complex, further investigations of type specimens representing all synonyms (or topotypes, in case of old type specimens or those not preserved in ethanol) are needed by implementing DNA taxonomy together with morphological investigation.
In summary, the global scale phylogeny and species delimitation of the cosmopolitan littoral earthworm P. litoralis were here investigated by an integrative taxonomic approach, with both single and multi-locus multispecies coalescent-based species delimitation methods. The study revealed several MOTUs within P. litoralis based on COI species delimitation alone, and this was well supported by the ITS2 data. The phylogenetic tree shows deeply divergent mitochondrial lineages and a high number of haplotypes, especially for COI. Without support from morphological characteristics, we suggest that the morphospecies P. litoralis is referred to as a cryptic species. Further in-depth studies of the morphology and anatomy of these littoral earthworms, e.g., by using scanning electron microscopy, are required to investigate the potential presence of cryptic morphology, which would provide further evidence for a more precise taxonomic revision of the species complex. Moreover, studies on population genetics and a search for more evidence (or lack) of gene flow and/or reproductive barriers are needed.
Materials and methods
Specimen collection and morphological examination
Specimens of P. litoralis were collected from several types of habitats, such as sandy beaches, mangrove swamps of the intertidal zone, sanitary sewer links, estuaries, under the trash or leaf litter, and freshwater channels between the mainland and the sea, in Thailand and surrounding countries in Southeast Asia (Fig. 1A,B) since 2007. All specimens were deposited in Chulalongkorn University Museum of Zoology (CUMZ), Thailand. Additional Japanese, Taiwanese, and Fijian specimens deposited in the collection at Chubu University Japan were included in the analyses. These littoral earthworms could be found in sand mixed with seaweed debris in sandy beaches facing the ocean in Taiwan and Japan, ranging from the northernmost record at Matsushima Bay, Miyagi Prefecture, to Aichi Prefecture, Mideast Honshu, Fukuoka Prefecture, Kyushu, and the Ryukyu archipelago. In addition, additional specimens of P. litoralis were collected by Christer Erséus and his team from different beaches at Lizard Island (Great Barrier Reef, Australia), Carrie Bow Cay (the barrier reef of the coast of Belize), and from three localities in Southeastern USA: Cedar Point (Alabama), Craig Key (Florida Keys), and Indian River Lagoon at Fort Pierce (Florida), the two latter sites being about 350 km apart. The Australian sites were all in depressions immediately behind the beach sand, while the US and Belizean sites were in the upper intertidal zone on the seaward slope of the beach. Finally, worms were also obtained from Turkey (Biga Peninsula in Marmara Sea; courtesy of Sermin Acik Cinar) and South Africa (Grahamstown; courtesy of Sam James). All specimens were preserved in 80–99% (v/v) ethanol for molecular analyses. For other details of the worms used in the analysis, see Table 1. Morphological identification (Fig. 1C) was made based on taxonomic literature following Easton (1984), Gates (1972), and Seesamut et al., (2018)1,2,30. All work with animals was conducted in accordance with the Institutional Animal Care and Use Committee of Khon Kaen University (IACUC-KKU) under approval number IACUC-KKU-32/65.
DNA extraction, PCR amplification, and DNA sequencing
Voucher specimens of P. litoralis from Southeast Asia and Japan, including Taiwan, were used for the extraction of their total genomic DNA from the posterior part of each earthworm using the Lysis Buffer for PCR (Takara) and following the manufacturer's protocol. Two molecular markers were amplified: a fragment of mitochondrial COI and the internal transcribed spacer 2 (ITS2) region of the nuclear ribosomal DNA. The COI fragment was amplified with the Tks Gflex™ DNA Polymerase (Takara) using universal primers HCO2198 and LCO 149039, while primers 606F (forward) and 1082R (reverse)40 were used for ITS2. The PCR mixture was as follows: 1 μL of Tks Gflex DNA Polymerase (1.25 unit/μL), 25 μL of 2 × Gflex PCR buffer (Mg2+, dNTP plus), 1 μL each of primers (10 μM), 19.5 μL of sterilized distilled water, and 2.5 μL of crude lysate with Lysis buffer. The PCR thermal cycling was performed as 94 °C for 2 min, followed by 35 amplification cycles of 94 °C for 60 s, 48 °C for 60 s, and 72 °C for 2 min and then followed by a final 72 °C for 5 min. The concentration and quality of the amplicons were examined by 1% (w/v) agarose gel electrophoresis against a DNA standard marker in 1 × TAE buffer and detected under UV transillumination after staining with SYBR® Safe DNA Gel Stain. The samples for which direct sequencing of the nuclear gene markers failed were subjected to subcloning using Promega pGEM-T Easy Vector System (Promega, Cat: A1360) to separate allelic variants before sequencing. The purifying and sequencing of PCR products were done commercially by Macrogen Inc. (Japan).
For the specimens from the remaining localities, DNA was extracted from small pieces of worm tissue with the E.Z.N.A.® Tissue DNA Kit II (Omega Bio-tek), following the instructions for kits requiring OB protease, or in some cases with DNeasy® Blood & Tissue Kit (250) (QIAGEN). For samples extracted with E.Z.N.A., the tubes were incubated at room temperature for five minutes before eluting the DNA. The remaining parts of the specimens were deposited, as vouchers, in the Swedish Museum of Natural History, Stockholm. The extracted DNA was then used to PCR amplify fragments of the COI gene and nuclear ITS2 region using puReTaq Ready-To-Go PCR Beads (GE Healthcare). Amplification was done according to the kit instructions. The COI sequences were amplified by thermal cycling with an Eppendorf PCR, programmed at 35 cycles of 40 s at 95 °C, 45 s at 45 °C, and 1 min at 72 °C, with an initial denaturation period of 5 min at 95 °C and a final terminal extension period of 8 min at 72 °C. For the ITS region, there were 25 cycles of 30 s at 95 °C, 30 s at 50 °C and 1 min at 72 °C with the same denaturation and extension period as for COI. The PCR products were checked with electrophoresis on agarose gel (1%) stained with ethidium bromide (3%), and the successfully amplified PCR products were purified using an E.Z.N.A® Cycle-Pure Kit (GE Healthcare) according to the manufacturer’s instructions, except for 100 μL of CP buffer was used and the final elution was done with 40 μL sterile deionized water. The products were then sent to Macrogen Inc., South Korea, where all samples were sequenced.
Sequence editing, alignment, phylogenetic reconstruction, and haplotype analysis
To identity and verify the amplified sequences, the obtained sequences were submitted to the BLASTn algorithm to check and compare with other sequences available in the GenBank databases in the National Center for Biotechnology Information- NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). All sequences were reassembled, edited, and aligned in MEGA X41 using the MUSCLE algorithm42 with default parameters, and then manually checked by eyes.
The phylogenetic analyses of the COI gene and the concatenated dataset (COI + ITS2) were conducted. The best-fit nucleotide substitution model of each gene fragment for phylogenetic analysis was determined using JModelTest v2.1.1043. Phylogenetic trees were reconstructed under maximum likelihood (ML) through the online portal CIPRES Science Gateway44 as implemented in RaxML-HPC2 on XSEDE45, with 1,000 bootstrapping replicates and default parameter settings. The ML tree based on the RaxML program was constructed under the GTR + CAT model for the best-fit nucleotide substitution. The resulting tree was plotted using FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree) and the tree diagram was created in Adobe Photoshop 2020. The ML analysis of the concatenated data (COI and ITS2) was done after partitioning the concatenated data with Kakusan446. For the haplotype analysis, the NEXUS format was created by DnaSP v.647 and the haplotype networks were constructed in PopArt48 using the TCS method49. Genetic divergences were examined using uncorrected p-distance as implemented in MEGA X with a bootstrap re-analysis of 1,000 pseudoreplicates.
Mitochondrial and multi-locus species delimitation analyses
Molecular species delimitation using the COI sequences was performed using the ABGD20, ASAP21, bPTP22 and GMYC23 methods. The ABGD is a simple method to split a sequence alignment data set into candidate species. We used the ABGD online server with default settings, to divide the specimens into clusters (http://wwwabi.snv.jussieu.fr/public/abgd). The ASAP analysis21 was implemented in an online web server (https://bioinfo.mnhn.fr/abi/public/asap/) under Kimura (K80) model. The lowest score was considered50. The bPTP analysis was carried out using an online web server (https://species.h-its.org/) with 100,000 MCMC generations. The GMYC method is a likelihood method for delimiting species by fitting within- and between-species branching models to reconstruct gene trees. The initial Bayesian tree was constructed in the BEAST v1.10.4 package51,52. All parameter settings were configured in BEAUTi v1.8.4, while Tracer v1.6 was used to check the estimate sample size (ESS) values and run the trace file. Using the ultrametric tree produced by BEAST, the GMYC analysis was performed in the R package splits.
Multi-locus species delimitation was performed using BPP v.3.328,29 on the COI and ITS2 datasets used in the ML analysis. The molecular operational taxonomic units (MOTUs) obtained from the GMYC analysis was used as the input as this analysis yielded the highest number of MOTUs, except for one MOTU for which no ITS2 sequence was available and so this MOTU was omitted from the analysis. The joint Bayesian species delimitations and species tree estimations28,53,54 were used, and three analyses (A-C) with different population size (estimated by θ) and divergence time (τ0) priors were performed, using the same settings and priors as in Martinsson and Erséus (2018)55 and Martinsson et al., (2020)32 (A: θ = 2, 400, τ0 = 2, 200; B: θ = 2, 1000, τ0 = 2, 200; C: θ = 2, 2000, τ0 = 2, 200). Each analysis was run for 200,000 generations, discarding the first 4,000 as burn-in, and all analyses were performed three times to confirm consistency between runs. We considered the species delimited with a PP (posterior probability) > 0.95 in all analyses to be well supported.
Data availability
Correspondence and requests for materials should be addressed to C.E. or S.P.
References
Easton, E. G. Earthworms (Oligochaeta) from islands of the south-western Pacific, and a note on two species from Papua New Guinea. N. Z. J. Zool. 11, 111–128 (1984).
Seesamut, T., Sutcharit, C., Jirapatrasilp, P., Chanabun, R. & Panha, S. Morphological and molecular evidence reveal a new species of the earthworm genus Pontodrilus Perrier, 1874 (Clitellata, Megascolecidae) from Thailand and Peninsular Malaysia. Zootaxa 4496, 218–237 (2018).
Seesamut, T., Jirapatrasilp, P., Chanabun, R., Oba, Y. & Panha, S. Size variation and geographical distribution of the luminous earthworm Pontodrilus litoralis (Grube, 1855) (Clitellata, Megascolecidae) in Southeast Asia and Japan. Zookeys 862, 23–43 (2019).
Seesamut, T. et al. Occurrence of bioluminescent and nonbioluminescent species in the littoral earthworm genus Pontodrilus. Sci. Rep. 11, 8407 (2021).
Grube, E. Beschreibungen neuer oder wenig bekannter Anneliden. Arch. Naturgesch 21, 81–136 (1855).
Perrier, E. Sur un nouveau genre indigene des Lombriciens terrestres (Pontodrilus marionis EP). Comptes rendus hebdomadaires des séances de ľAcadémie des sciences Paris 78, 1582–1586 (1874).
Beddard, F. E. A Monograph of the Order Oligochaeta (Clarendon Press, 1895).
Chen, S.-Y., Hsu, C.-H. & Soong, K. How to cross the sea: Testing the dispersal mechanisms of the cosmopolitan earthworm Pontodrilus litoralis. R. Soc. Open Sci. 8, 202297 (2021).
Blakemore, R. J. Origin and means of dispersal of cosmopolitan Pontodrilus litoralis (Oligochaeta: Megascolecidae). Eur. J. Soil Biol. 43, S3–S8 (2007).
Seesamut, T., Ng, B., Sutcharit, C., Chanabun, R. & Panha, S. Responses to salinity in the littoral earthworm genus Pontodrilus. Sci. Rep. 12, 22304 (2022).
Hebert, P. D., Cywinska, A., Ball, S. L. & deWaard, J. R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 270, 313–321 (2003).
Huang, J., Xu, Q., Sun, Z. J., Tang, G. L. & Su, Z. Y. Identifying earthworms through DNA barcodes. Pedobiologia 51, 301–309 (2007).
Chang, C. H., Rougerie, R. & Chen, J. H. Identifying earthworms through DNA barcodes: Pitfalls and promise. Pedobiologia 52, 171–180 (2009).
Jeratthitikul, E., Bantaowong, U. & Panha, S. DNA barcoding of the Thai species of terrestrial earthworms in the genera Amynthas and Metaphire (Haplotaxida: Megascolecidae). Eur. J. Soil Biol. 81, 39–47 (2017).
Hussain, M. et al. DNA barcoding: Molecular identification and phylogenetic analysis of pheretimoid earthworm (Metaphire sp. and Amynthas sp.) based on mitochondrial partial COI gene from Sialkot. Pak. J. Oleo Sci. 71, 83–93 (2022).
Pérez-Losada, M., Eiroa, J., Mato, S. & Domínguez, J. Phylogenetic species delimitation of the earthworms Eisenia fetida (Savigny, 1826) and Eisenia andrei Bouché, 1972 (Oligochaeta, Lumbricidae) based on mitochondrial and nuclear DNA sequences. Pedobiologia 49, 317–324 (2005).
Martinsson, S. & Erséus, C. Cryptic speciation and limited hybridisation within Lumbricus earthworms (Clitellata: Lumbricidae). Mol. Phylogenet. Evol. 106, 18–27 (2017).
Bozorgi, F. et al. Multigene phylogeny reveals a new Iranian earthworm genus (Lumbricidae: Philomontanus) with three new species. PLoS ONE 14, e0208904 (2019).
Jirapatrasilp, P., Backeljau, T., Prasankok, P., Chanabun, R. & Panha, S. Untangling a mess of worms: Species delimitations reveal morphological crypsis and variability in Southeast Asian semi-aquatic earthworms (Almidae, Glyphidrilus). Mol. Phylogenet. Evol. 139, 106531 (2019).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877 (2012).
Puillandre, N., Brouillet, S. & Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 21, 609–620 (2021).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876 (2013).
Fujisawa, T. & Barraclough, T. G. Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 62, 707–724 (2013).
Martinsson, S. & Erséus, C. Cryptic Clitellata: Molecular species delimitation of clitellate worms (Annelida): An overview. Diversity 13, 36 (2021).
Goulpeau, A. et al. OTU delimitation with earthworm DNA barcodes: A comparison of methods. Diversity 14, 866 (2022).
Brito, P. H. & Edwards, S. V. Multilocus phylogeography and phylogenetics using sequence-based markers. Genetica 135, 439–455 (2009).
Li, F. et al. Multilocus species delimitation and phylogeny of the genus Calommata (Araneae, Atypidae) in southern China. Zool. Scr. 51, 199–216 (2022).
Yang, Z. & Rannala, B. Unguided species delimitation using DNA sequence data from multiple loci. Mol. Biol. Evol. 31, 3125–3135 (2014).
Yang, Z. The BPP program for species tree estimation and species delimitation. Curr. Zool. 61, 854–865 (2015).
Gates, G. E. Burmese earthworms, an introduction to the systematics and biology of megadrile oligochaetes with special reference to South-east Asia. Trans. Amer. Philos. Soc. 62, 1–326 (1972).
Shekhovtsov, S. V., Golovanova, E. V. & Peltek, S. E. Cryptic diversity within the Nordenskiold’s earthworm, Eisenia nordenskioldi subsp. nordenskioldi (Lumbricidae, Annelida). Eur. J. Soil Biol. 58, 13–18 (2013).
Martinsson, S., Klinth, M. & Erséus, C. Testing species hypotheses for Fridericia magna, an enchytraeid worm (Annelida: Clitellata) with great mitochondrial variation. BMC Evol. Biol. 20, 116 (2020).
Buckley, T. R. et al. Phylogenetic analysis of New Zealand earthworms (Oligochaeta: Megascolecidae) reveals ancient clades and cryptic taxonomic diversity. Mol. Phylogenet. Evol. 58, 85–96 (2011).
Porco, D. et al. A reference library of DNA barcodes for the earthworms from Upper Normandy: Biodiversity assessment, new records, potential cases of cryptic diversity and ongoing speciation. Appl. Soil Ecol. 124, 362–371 (2018).
Marchán, D. F., DíazCosín, D. J. & Novo, M. Why are we blind to cryptic species? Lessons from the eyeless. Eur. J. Soil Biol. 86, 49–51 (2018).
Lohse, K. Can mtDNA barcodes be used to delimit species? A response to Pons et al. 2006. Syst. Biol. 58, 439–442 (2009).
Sukumaran, J. & Knowles, L. L. Multispecies coalescent delimits structure, not species. PNAS 114, 1607–1612 (2017).
Klein, A., Eisenhauer, N. & Schaefer, I. Invasive lumbricid earthworms in North America-Different life histories but common dispersal?. J. Biogeogr. 47, 674–685 (2020).
Folmer, O., Back, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Liu, Y. & Erséus, C. New specific primers for amplification of the Internal Transcribed Spacer region in Clitellata (Annelida). Ecol. Evol. 7, 10421–10439 (2017).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE) (New Orleans, LA, 2010).
Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Tanabe, A. S. Kakusan4 and Aminosan: Two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol. Ecol. Resour. 11, 914–921 (2011).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Leigh, J. W. & Bryant, D. Popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Clement, M., Posada, D. & Crandall, K. A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Ballarin, F. & Eguchi, K. Integrative taxonomic revision of the genera Nesticella and Howaia in Japan with the description of five new species (Araneae, Nesticidae, Nesticellini). ZooKeys. 1174, 219–272 (2023).
Drummond, A. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Yang, Z. & Rannala, B. Bayesian species delimitation using multilocus sequence data. PNAS 107, 9264–9269 (2010).
Rannala, B. & Yang, Z. Improved reversible jump algorithms for Bayesian species delimitation. Genetics 194, 245–253 (2013).
Martinsson, S. & Erséus, C. Cryptic diversity in supposedly species-poor genera of Enchytraeidae (Annelida: Clitellata). Zool. J. Linn. Soc. 183, 749–762 (2018).
Acknowledgements
This work was supported by Thailand Research Fund (TRF-DPG628001), Center of Excellence on Biodiversity (BDC-PG2-161002), and the National Research Council of Thailand (N35E660138). Authors give thanks and appreciation to all members of the Animal Systematics Research Unit, Chulalongkorn University (ASRU) for assisting in the fieldwork. Thanks also go to Hua-Te Fang for the specimens from Taiwan. We are also grateful to Sermin Acik Cinar and Sam James for providing material.
Author information
Authors and Affiliations
Contributions
Conceptualization, T.S. and S.P.; fieldwork, T.S., Y.O., M.L., C.E. and S.P.; methodology, T.S., P.J. and M.L; investigation, T.S., P.J., S.M. and M.L.; writing original draft preparation, T.S.; writing, review and editing, T.S., Y.O., P.J., S.M., C.E. and S.P.; supervision, C.E. and S.P. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seesamut, T., Oba, Y., Jirapatrasilp, P. et al. Global species delimitation of the cosmopolitan marine littoral earthworm Pontodrilus litoralis (Grube, 1855). Sci Rep 14, 1753 (2024). https://doi.org/10.1038/s41598-024-52252-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52252-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
