
Abstract
The golden jackal (Canis aureus) is a reoccurring species in the centre of the Carpathian basin, in Hungary. In total, 31 golden jackal tissue samples were collected, from 8 white-coated, 2 black-coated and one mottled animal across Hungary. Sequences and fragment length polymorphisms were studied for white colour (MC1R), and for black coat colouration (CBD103). In each white animal, the most widespread mutation causing white fur colour in dogs in homozygous form was detected. Three animals were found to carry the mutation in heterozygous form. The two black golden jackals were heterozygous for the 3 bp deletion in CBD103 that mutation for black coat colouration in dogs, and one of them also carried the mutation causing white fur. None of the white animals showed signs of hybridization, but both the black and the mottled coloured individuals were found to be hybrids based on genetic testing. Kinship was found three times, twice between white animals, and once between a white animal and an agouti animal carrying the mutation of white coat. Our results confirm the findings that golden jackal–dog hybrids may occur without human intervention, and the detected mutation causing white fur colour in golden jackals could possibly be due to an early hybridization event.
Similar content being viewed by others


Introduction
The golden jackal (Canis aureus) is a medium-sized canine leading an opportunistic lifestyle1. Its distribution is mainly across Northern Africa, Southern Europe, Near East, India and South Asia, but it is rapidly spreading towards Northern and Western European areas2. In Eastern Europe and the Balkan region its presence was continuous during the twentieth century and its numbers were constant3, but in Hungary during the 19th and the majority of the twentieth century only stray animals were detected4,5,6. The expansion of the species started from Bulgaria in the 1960s7; soon its numbers started to rise in Romania8, and there were recordings of golden jackals in Serbia at the end of the 1970s–beginning of 1980s5. In Hungary, the first observations of relocation occurred at the beginning of the 1980s5, and ever since, its numbers are rising9 despite local population management, like trapping and extensive hunting. The golden jackal is generally found in open grasslands, but with urbanization and its opportunistic habitat use, jackals can now be found around human settlements as well10.
Genetically, the golden jackal is a close relative of the coyote (Canis latrans), the grey wolf (Canis lupus), the African wolf (Canis lupaster), the African golden wolf (Canis anthus) and the dog (Canis familiaris)11. Phylogenetic and evolutionary studies show that wolves, coyotes and dogs share genomic elements12, and there is significant gene flow between different Canid species13,14,15.
In North America, hybridization between the different Canid species has been observed and well documented, and it has been found recently that grey wolves, red wolves, coyotes and Algonquin wolves show little to no genomic differences16,17. Even though most wolf species fall under the Endangered Species Act of the USA18, there have been several attempts to establish wolf-dog hybrid breeds, such as the Calupoh19.
Coyotes are known to interbreed with dogs20,21, and genetic analyses of mitochondrial DNA show that dog ancestry is widespread in coyotes in the southeastern United States14. In 2010, Kays et al.22 found that eastern coyotes interbreed with wolves in the northeastern area of the US. They also found that this hybridization contributed to a larger body size, which resulted in better hunting abilities regarding deer, and the larger coyotes filled a vacant niche in the area left by the eradication of wolves15,23. These coyote-wolf hybrids are generally known as coywolves24, but whether they should be considered a subspecies is still heavily debated22.
Hybridization with dogs has caused several phenotypic changes in wolves, such as black coat colouration in North American grey wolves, which was introduced to the species at least 7500 years ago through introgression with dogs25, but there have also been documented cases of hybridization between dogs and coyotes25 and wolves in North America26 resulting in several morphological changes.
There is no direct evidence of hybridization between grey wolves (Canis lupus) and golden jackals in the wild, but there is genetic evidence of admixture between golden jackals and African wolves (Canis lupaster) in Israel27.
Historical anecdotes28,29 can be found about possible jackal–dog hybrids. Erik Ziman described breeding experiments with poodles and jackals30, and well-documented evidence for human-made dog–jackal hybrids are the Sulimov dogs31,32. Recently in India, camera trap pictures have shown golden jackals showing dog-like pelt colouration in urban areas33, which could be due to possible hybridization. One documented instance of hybridization in the wild was confirmed by genetic studies, which was described by Galov et al. in34.
Phenotypic changes resulting in pheomelanistic coat due to MC1R mutations are observed in many mammal species35,36,37, but a loss-of-function mutation is also responsible for the white or golden colouration of dogs’ fur colour38. The same mutation could also be found in coyotes39 in North America.
In dogs, there are two widespread MC1R mutations: the first is the dominant Em (melanistic mask), which is caused by a valine to methionine change at amino acid 264, resulting in a melanistic mask on the face, dark underside and legs40. The other is a “recessive yellow” allele “e”, which changes an arginine to a premature stop codon at amino acid position 306, and is considered recessive38, therefore can remain hidden for generations without selection41. Breed-specific mutations exist in sighthounds42, English Cocker Spaniels, huskies and Australian cattle dogs43, but they have not been found in wild canids yet.
Black or melanistic coat colour in dogs is caused by a 3-basepair in-frame deletion (ΔG23) in the canine beta-defensine 103 gene (CBD103)44,45. This ΔG23 has been found in North American wolves25, Italian wolves46, coyotes47, and in golden jackal-dog hybrids described by Galov et al.34. Melanism is considered a dominant trait, therefore one copy of the mutation is enough to create a phenotypic change with a properly working melanocortin-1 receptor.
Merle coat colouration in dogs is caused by the insertion of a short interspersed element (SINE) into the SILV gene and results in diluted areas of eumelanistic fur, showing a mottled phenotype in heterozygous form48,49, whereas in homozygous form it associates with ocular abnormalities and deafness50. Clark et al. reported that several dogs with non-merle phenotype were heterozygous for a smaller insertion, and Langevin et al. found that length differences in the poly-A tail of the SINE insertion in the SILV gene results in differences in the merle phenotypes of dogs that can be categorized into six groups based on the poly-A tail length (Mc, Mc +, Ma, Ma +, M, Mh)51.
Recently there have been several documented and legally shot white-coloured golden jackals in the southern areas of Hungary, as well as black and mottled animals. Our aim in this study was to determine the mutations behind their fur colour, and to detect whether the change in their phenotype was due to possible hybridization with dogs, using microsatellite markers and mitochondrial sequencing.
Results
MC1R results
As a reference sequence we used the 954 bp long MC1R sequence of ROS_Cfam_1.0 [Accession number: NC_051809.1:c63923224-63922271].
Four SNPs were present in our samples, c.475C>T, c.476C>A, c.790A>G and c.916C>T; the latter of these is the stop codon mutation for white coat colouration in dogs.
At c.475, where most agouti-coloured golden jackals are homozygous for the T allele, which results in a proline to serine amino acid change, one animal showed heterozygosity. At c.476, white-coloured jackals are all homozygous for the A allele, resulting in a proline to glutamine amino acid change. We found two instances of heterozygosity of c.790A>G, which is the mutation causing masked phenotype in dogs. At c.916, all white animals (Fig. 1) were homozygous for the T allele, which results in a stop codon terminating the open reading frame of the melanocortin receptor gene. We also found three animals heterozygous for the c.916C>T mutation.

Photographs of the white animals, 4744CaW (a), 4798CaW (b), 6392CaW (c), 6350CaW (d), 6343CaW (e), 6647CaW (f), 6648CaW (g), 6651CaW (h).
Analysing the other previously described SNPs in the MC1R sequences38, at Ser90Gly (c.268) all of our samples were homozygous for the A allele, at Ala105Thr (c.313) they were all homozygous for the G allele, and at Arg301Cys (c.901) all samples were homozygous for the C allele (Supplementary Table 1).
CBD103 results
Not only black, but also white and mottled jackals were genotyped for the ΔG23 mutation of the CBD103 gene, since the 916C>T MC1R mutation is epistatic in homozygous form to this mutation. Using fragment analysis, we found that the black animals (Fig. 2) were heterozygous for the 3-basepair deletion, and all other animals were homozygous for the wild-type allele (Supplementary Table 1).

The black animals (5920HybB, 5928HybB) that are heterozygous for the CBD103 deletion.
SILV results
In the mottled animal’s case (Fig. 3), the phenotype was highly resemblant to agouti or dark sable merle dogs’ look that is caused by a SINE insertion in the SILV gene. Using fragment analysis, we found a peak at 434 basepairs with a wild-type allele of 171 basepairs, which means the animal has a 263-basepair SINE insertion, which categorizes it as Ma +, atypical merle animal.

The animal (5930HybM) showing merle phenotype and carrying a SILV SINE insertion.
Partial mitochondrial DNA sequencing
We analysed the mitochondrial D-loop sequence of the 31 animals acquired for this research using BLAST. In 30 cases the results showed golden jackal origins; sample 5928HybB, however, was identified as a dog (GenBank Accession number: OR047611), which indicates possible dog maternity.
Haplotype analysis revealed that out of the 31 animals in this study, 30 belong to the same haplogroup, with the exception of the animal with dog mitochondria. (Supplementary Table 1). This result confirms the previous findings that golden jackals show a high degree of genetic uniformity52.
Microsatellite analysis, structure and kinship results
All loci were polymorphic, with the mean number of alleles reaching 5.52, with average expected- and observed heterozygosity being 0.682 and 0.660, respectively (Supplementary Tables 2, 3, 4), all loci were therefore included in subsequent multi-locus analyses. Using 20 polymorphic microsatellite markers, we determined the genetic distances of our samples from dog and golden jackal samples. Structure results showed possible F1 hybridization in three cases (Fig. 4).

Result of the STRUCTURE analyses at K = 2. Group 1 = reference dogs (n = 23), Group 2 = reference jackals (n = 27), Group 3 = agouti jackals (n = 20), Group 4 = white jackals (n = 8), Group 5 = Black and mottled animals (n = 3). The Delta K value shows the most likely number of clusters, which is 2 in this case. The colour red indicates clustering with the dog group; green is the colour of the golden jackal cluster. In group 5, the individuals show approximately 50% clustering to both groups, so they cannot be clustered into either.
At K = 2, the reference populations separated completely, and all animals in the agouti and white group belonged to the jackal population. The two black and the mottled jackals could not be assigned to the reference groups with an assignment probability of 0.15 and 0.85.
Principal Coordinates Analyses show that the white animals are in the same genetic cluster as the agouti-coloured jackals, whereas the black and mottled animals fall between the dogs and golden jackals (Fig. 5).

Principal coordinates analysis of the animals in this study. Yellow = reference dogs, pink = reference jackals, red = agouti group, green = white group, blue = black and mottled animals. The white animals are all grouped together with the reference and agouti groups, whereas the coloured animals fall between the reference dog and jackal groups.
Using STRUCTURE for clustering analysis, we found that none of the artificial genotypes in the F1, F2, and F3 groups could be clustered into either parental cluster. Supplementary Table 5 and Fig. 6 show the results of the artificial genotypes clustering analyses.

Clustering analysis of the artificial genotypes created by Hybridlab. Group 1: Parental dog (n = 27), Group 2: Parental golden jackal (n = 27), Group 3: F1 (CfxCa) (n = 10), Group 4: F1xDog backcross (n = 10), Group 5: F1xJackal backcross (n = 10), Group 6: F2 (F1xF1) (n = 10), Group 7: F2xDog backcross (n = 10), Group 8: F2xJackal backcross (n = 10), Group 9: F3 (F2xF2) (n = 10). Delta K value shows the most likely number of clusters, which is 2 in this case.
We performed kinship analysis with monogamous settings, to determine whether there is a relationship between the white, white carrying, and hybrid animals (n = 13). We found three family clusters between the white and white carrying animals with one likely (Probability > 0.75, 6647CaW/6648CaW/6651CaW) sibling group and two possible sibling groups (Probability > 0.5, 4744CaW/6343CaW and 4802CaA/6392CaW) (Supplementary Table 6).
One cluster (6647CaW/6648CaW/6651CaW) showed close sibling or parent–offspring relationship, which was confirmed by the place and time of the catch—all animals were the same age and killed in the same den at the same time.
Discussion
With the recent resettlement of golden jackals in the Hungarian lowland, where intensive agricultural involvement and stray or free-ranging dogs are present in areas with low population density, jackals have many opportunities for admixture with local dogs. Since hybridization has not been studied in this area, our research highlights the possibility that there might have been possible earlier hybridization events between dogs and golden jackals.
Analysing the MC1R gene of the animals in this study, we found that the white animals (n = 8) are all homozygous for the R306Ter (c.916C>T) mutation that results in an early stop codon, leading to an entirely pheomelanistic, white colouration, but they are also homozygous for the c.476C>A, a proline to glutamine change. Compared to MC1R sequences downloaded from the NCBI database, we found that the SNP has been described by Wang et al.53, and it is present in the Kunming Dog population, as well as in dog breeds from FCI Group 7., Ancient breeds: Alaskan Malamutes, Siberian Huskies, Shar peis, Chowchows, Akitas, Eurasiers, Tibetan mastiffs, it was also found in salukis, beagles and chihuahuas54, but it could not be found in other publicly available dog, wolf and coyote MC1R sequences. Since mainly ancient type dogs carry the same mutations as our white samples, it is possible that the “e” allele entered the gene pool through an earlier hybridization event with an ancient type dog.
We also found an additional SNP in the MC1R sequences: at c.475 there is a C>T, proline to serine amino acid change, which appeared only in agouti jackals, and was not found it in the dog, wolf, and coyote MC1R sequences available in the NCBI database.
Galov et al. in34 found several wild golden jackal-dog hybrids and confirmed their hybrid status through genetic evidence, supporting the assumption that dogs and golden jackals are capable of interbreeding in areas, where their territories overlap. Stronen et al. proved16 that human agricultural intervention can induce hybridization in otherwise sympatric species. Our findings also confirm that hybridization naturally occurs between jackals and dogs, and phenotypic changes can be expected, the most spectacular being the black coat colouration caused by a deleterious mutation in CBD103 originating from dogs, that was found by both Galov et al. and ourselves.
Since the hybrid animals in our study are not relatives, we can here report three different hybridization events, which raises the question how often it happens, and what happened to the littermates of the animals, whether they were unreportedly shot or they are still in the breeding population. The case of the mottled, merle animal sheds light on the dangers of hybridization with dogs—longer SILV SINE insertions in homozygous form can cause developmental issues, like hearing loss and micropthalmia55, and since dogs carry at least 152 possible genetic health issues56, most of them recessive, developing symptoms after reaching sexual maturity, they are capable of hiding in the gene pool for generations, leading to possible regression of health in isolated populations.
Barash et al.27 found evidence for possible hybridization of golden jackals with dogs in Israel that confirms our findings of naturally occurring dog-jackal hybrids, and found introgression with African wolves (Canis lupaster), which shows that golden jackals are prone to interbreeding with other Canid species, where their habitats overlap. In Europe, besides free roaming dogs, the only other Canid species golden jackals might share a habitat with are grey wolves (Canis lupus), but wolves possibly have a key role in the regulation of the golden jackal population3, and we have found no evidence of hybridization between the two species.
Coyotes’ and wolves’ hybridization with dogs has already raised questions regarding the behaviour of the introgressed populations—they become bolder and possibly more dependent on human resources57; however, the effects of hybridization on the opportunistic lifestyle of golden jackals is still unknown and needs more investigation.
In conclusion, we identified the mutation responsible for white coat in golden jackals, which is the same as in dogs. This allele was introduced to the species possibly through an earlier hybridization event with an ancient type dog. We also identified three dog-golden jackal hybrid animals, two of them are heterozygous for the CBD103 ΔG23 mutation causing black coat, and one of them is heterozygous for the SILV SINE insertion causing merle pattern in dogs. Our findings confirm that golden jackals interbreed with dogs and hybridization with dogs results in phenotypic change, which in recessive form can stay hidden for generations. A recessive allele for a hereditary disease entering the population from the dogs’ side could pose a danger for isolated jackal populations.
Methods
Sampling and genomic DNA extraction
Agouti (n = 20), white (n = 8), black (n = 2) and mottled (n = 1) golden jackal (Fig. 7) muscle and liver tissue samples were collected from legally hunted, free-ranging animals between 2014 and 2021 at hunting sites localized in the southern regions of Hungary (Somogy county, Baranya county, Csongrád-Csanád county) (Figs. 8, 9).

Examples of the coat colourations of the jackals in this study. From left to right: Agouti (sample 6339CaA), white (sample 4744CaW), black (sample 5920HybB) and mottled (sample 5930HybM).

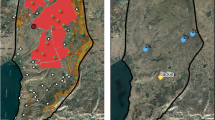
Sampling locations: 1—Inner Somogy Hunting Region (n = 19), 2—South Balaton Hunting Region (n = 1), 3—Zselic—Mid-Somogy Hunting Region (n = 2), 4—Mecsek Hunting region (n = 2), 5—Illancs-Bugac Hunting Region (n = 2), 6—Maros-Csongrád Hunting Region (n = 2). A Austria, SLO Slovenia, CRO Croatia, SRB Serbia, RO Romania, UA Ukraine, SK Slovakia, HUN Hungary.

Sample collection sites of the coloured animals. CRO Croatia, SRB Serbia, RO Romania.
As reference samples for studying hybridization levels, hair with follicles (n = 27) from legally shot, free-ranging golden jackals from observed populations, and samples from mixed breed and pedigree dogs (n = 23) were collected and stored at − 20 °C until further laboratory procedure.
Genomic DNA isolation was performed using the MagCore HF16 Automated Nucleic Extractor® with the MagCore Genomic DNA Tissue Kit® (RBC Bioscience Corp., Taiwan), according to the manufacturer’s protocol; the genomic DNA samples were stored at − 20 °C.
Genetic analysis of colour
MC1R sequencing was performed with D and E primers described by Newton et al.38 (primer D: 5′-GGTCATTGCTGAGCTGACAC-3′, primer E: 5′-GAGATGCTGTCCAGTAGTCCC-3′). The volume of the PCR mixture was 25 µl, using Multiplex MasterMix (Qiagen Gmbh, Germany); 0.4 µl of each primer diluted to 10 µM, and 120 ng of DNA diluted to 15 ng/µl. PCR was conducted in a GeneExplorer instrument (Hangzhou Bioer Technology Co., China). The PCR profile was the following: 95 °C for 15 min, 40 cycles of 94 °C for 30 s, 64 °C for 60 s, 72 °C for 2 min, followed by a final extension step of 72 °C for 5 min. The 1347-bp-long PCR products were purified using NucleoSpin® Gel and PCR Clean-up Kit (Macherey–Nagel Gmbh, Germany); Sanger-sequencing was executed using BigDye Terminator 3.1 Cycle Sequencing Kit and 3500xl Genetic Analyzer instrument (Applied Biosystems, USA). MC1R sequences were aligned using SeqMan Pro (DNAStar, Inc), and the alignments were checked manually. We used the MC1R sequence of ROS_Cfam_1.0 (NC_051809.1:c63923224-63922271, Labrador retriever) as a reference.
The MC1R sequences of the samples in this study are available under the Genbank accession numbers OR074050–OR074080.
Analysis of CBD103 was done using primers described by Leonard et al.44 (Forward: 5′-(6FAM) TCGGACCAAAGCCTTCCTAAAG-3′, Reverse: 5′-AACGCTCCCATCCCATTTCTGG-3′). The reaction mixture volume was 20 µl, it contained 4 µl of DNA diluted to the concentration of 15 ng/µl, 10 µl of Phire Animal Tissue PCR Buffer, 0.4 µl of Phire HS II DNA polymerase enzyme, 0.5 µl of each primer diluted to 10 µM and 6.6 µl nuclease-free water. PCR was conducted in a GeneExplorer instrument (Hangzhou Bioer Technology Co., China). The PCR profile was the following: 98 °C for 30 s, 35 cycles of 98 °C for 10 s, 55 °C for 10 s, 72 °C for 20 s, and final extension at 72 °C for 5 min. We expected two fragment lengths, the wild-type allele at 228 basepairs and the G23 mutation at 225 basepairs. The 3-basepair deletion in CBD103 was detected by fragment length analysis using Geneious Prime® (Biomatters Ltd).
Amplification of the SILV SINE insertion was done using the method and primers described by Langevin et al.51. We used Qiagen Multiplex MasterMix (Qiagen Gmbh, Germany) according to the producer’s instructions. The length of the SINE insertion was determined using Geneious Prime® (Biomatters Ltd).
Hybridization analyses
To detect possible hybridization with dogs and conclude kinship, we performed microsatellite and mitochondrial sequence analysis on the samples to detect the direction of hybridization.
Amplification of the hypervariable mitochondrial control region (D-loop) was performed with primers described by Fabbri et al.58 (5′-TCCCTGACACCCCTACATTC-3′, 5′-CGTTGCGGTCATAGGTGAG-3′) resulting in a 519-basepair amplicon. The reaction mixture volume was 20 µl, it contained 3 µl of DNA diluted to the concentration of 15 ng/µl, 10 µl of Phire Animal Tissue PCR Buffer, 0.4 µl of Phire HS II DNA polymerase enzyme, 0.6 µl of each primer diluted to 10 µM and 5.4 µl nuclease-free water. PCR was conducted in a GeneExplorer instrument (Hangzhou Bioer Technology Co., China). The PCR profile was the following: 98 °C for 30 s, 35 cycles of 98 °C for 20 s, 62.5 °C for 30 s, 72 °C for 70 s, and final extension at 72 °C for 5 min. PCR products were purified using NucleoSpin® Gel and PCR Clean-up kit (Macherey–Nagel Gmbh, Germany); Sanger-sequencing was executed using BigDye Terminator 3.1 Cycle Sequencing Kit and 3500xl Genetic Analyzer instrument (Applied Biosystems, USA).
For microsatellite analysis we amplified 20 loci in 4 multiplexed reactions using Qiagen Multiplex PCR Kit (Qiagen Gmbh, Germany): AHTH137, AHTH171, AHTH26059, FH2538, c2001, c2096, c2054, PEZ8, PEZ5, PEZ19, PEZ12, PEZ1160, FH3377, FH2088, FH3313, PEZ02, FH2010, FH2107, FH2004, FH230961.
The sex of the animals was identified by hunters and confirmed with the amelogenin gene62.
The reaction mixture volume was 25 µl, containing Multiplex MasterMix (Qiagen Gmbh, Germany), 120 ng of DNA, optimalized amounts of primers at 10 µM concentration, and nuclease-free water. PCR was conducted in a GeneExplorer instrument (Hangzhou Bioer Technology Co., China). The PCR profile for all multiplexes was the following: 96 °C for 15 min; 40 cycles of 95 °C for 30 s, 58 °C for 60 s, 72 °C for 60 s, and final extension at 60 °C for 30 min. Fragment capillary analysis was conducted on 3500xl Genetic Analyzer instruments with LIZ500 size standard (Applied Biosystems, USA).
The lengths of microsatellite fragments were described using Geneious Prime® (Biomatters Ltd) with Two Surrounding Peaks sizing method.
Mitochondrial sequences were aligned using SeqMan Pro (DNAStar, Inc), and the alignments were checked manually. For species identification, the sequences were loaded into the Basic Local Alignment Tool (BLAST®)63. Haplotype analyses were carried out using MEGA1164. The mitochondrial D-loop sequences of the samples in this study are available under the Genbank accession numbers OR047611–OR047641.
Statistical analysis
For genetic clustering we used STRUCTURE ver. 2.3.4.65; for analysis of STRUCTURE results, we used STRUCTURE Harvester v06.9466.
The Bayesian clustering method in STRUCTURE was used to ascertain the number of genetic clusters to define possible admixture of golden jackals and dogs. The Bayesian clustering method and the Markov Chain Monte Carlo (MCMC) simulation were run assuming no prior information and using an admixture and independent allele frequency models. The simulation was run with 7 independent runs for each K value ranging from 1 to 9, with a burn-in period of 250,000 iterations and 750,000 replications. The number of genetic clusters (K) was calculated with the second-order rate of change in log-likelihood values (ΔK) in STRUCTURE Harvester v06.9466.
Genetic distance analysis and Principal Coordinates Analysis were performed using GenAlEx 6.567.
To determine whether our microsatellite set is capable of distinguishing F1 or F2 hybrids, we created artificial genotypes using Hybridlab68 in the following iterations: parent clusters (dog and golden jackal), F1 (dog × jackal), dog and F1 hybrids, jackal and F1 hybrids, F2 (F1 × F1), dog and F2 hybrids, jackal and F2 hybrids and F3 (F2 × F2) hybrids. We created artificial parent populations with 27 genotypes each, and artificial offsprings with 10 genotypes each.
Kinship analysis was based on fragment length and was done using Colony v2.0.6.669 and Cervus 3.0.770.
Ethics statement
This study was out of the scope of the 2010/63/EU Directive, and was carried out with the knowledge and permission of the Institutional Animal Welfare Committee of Hungarian University of Agricultural and Life Sciences Szent István Campus (MATE-SZIC/1068-1/2023).
Data availability
The genetic sequences of the samples have been deposited in the public sequence repository NCBI GenBank, the mitochondrial control region (D-loop) sequences are available under accession numbers OR047611–OR047641, the MC1R sequences are available under the accession numbers OR074050–OR074080.
References
Lanszki, J. et al. Diet composition of the golden jackal in an area of intensive big game management. Ann. Zool. Fenn. 52, 243–255 (2015).
Kowalczyk, R., Wudarczyk, M., Wójcik, J. M. & Okarma, H. Northernmost record of reproduction of the expanding golden jackal population. Mamm. Biol. 100, 107–111 (2020).
Krofel, M., Giannatos, G., Ćirovič, D., Stoyanov, S. & Newsome, T. M. Golden jackal expansion in Europe: A case of mesopredator release triggered by continent-wide wolf persecution?. Hystrix Ital. J. Mammal. 28, 9–15 (2017).
Arnold, J. et al. Current status and distribution of golden jackals Canis aureus in Europe. Mamm. Rev. 42, 1–11 (2012).
Spassov, N. & Acosta-Pankov, I. Dispersal history of the golden jackal (Canis aureus moreoticus Geoffroy, 1835) in Europe and possible causes of its recent population explosion. Biodivers. Data J. 7, e34825 (2019).
Tóth, T., Krecsák, L., Szhucs, E., Heltai, M. & Huszár, G. Records of the golden jackal (Canis aureus Linnaeus, 1758) in Hungary from 1800th until 2007, based on a literature survey. North-West. J. Zool. 5, 386–405 (2009).
Nikolay, S. The position of jackais in the canis genus and iife-history of the goiden jackai (Canis aureus L.) in Buigaria and the baikans. Hist. Nat. Bulgar. 1, 44–56 (1989).
Kryštufek, B., Murariu, D. & Kurtonur, C. Present distribution of the Golden Jackal Canis aureus in the Balkans and adjacent regions. Mamm. Rev. 27, 109–114 (1997).
Szabo, L., Heltai, M., SzHucs, E., Lanszki, J. & Lehoczki, R. Expansion range of the golden jackal in Hungary between 1997 and 2006. Mammalia 73, 307–311 (2009).
Bulmer, L. (2015).
Koepfli, K.-P. et al. Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr. Biol. 25, 2158–2165 (2015).
Gopalakrishnan, S. et al. Interspecific gene flow shaped the evolution of the genus Canis. Curr. Biol. 28, 3441–3449 (2018).
Freedman, A. H. et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 10, e1004016 (2014).
Adams, J. R., Leonard, J. A. & Waits, L. P. Widespread occurrence of a domestic dog mitochondrial DNA haplotype in southeastern US coyotes. Mol. Ecol. 12, 541–546 (2003).
Kays, R., Curtis, A. & Kirchman, J. J. Rapid adaptive evolution of northeastern coyotes via hybridization with wolves. Biol. Lett. 6, 89–93 (2010).
Stronen, A. et al. Canid hybridization: Contemporary evolution in human-modified landscapes. Ecol. Evol. 2, 2128–2140 (2012).
von Holdt, B. M. et al. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2, e1501714 (2016).
United, S. The Endangered Species Act as amended by Public Law 97–304 (the Endangered Species Act amendments of 1982). (U.S. G.P.O., 1983).
. The Yucatan Times. Vol. 2022. (2019). www.theyucatantimes.com.
Young, S. P. & Jackson, H. H. T. The Clever Coyote (University of Nebraska Press, 1978).
Mengel, R. M. A study of dog-coyote hybrids and implications concerning hybridization in Canis. J. Mammal. 52, 316–336 (1971).
Kays, R. Vol. 2023 (Elsevier, 2015).
Way, J. G. & Lynn, W. S. Northeastern coyote/coywolf taxonomy and admixture: A meta-analysis. Canid Biol. Conserv. 19, 1–7 (2016).
Way, J. G. Vol. 2023 (The Conversation, 2016).
Anderson, T. M. et al. Molecular and evolutionary history of melanism in North American Gray wolves. Science 323, 1339–1343 (2009).
von Holdt, B. M. et al. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2, e1501714 (2023).
Barash, A. et al. Possible origins and implications of atypical morphologies and domestication-like traits in wild golden jackals (Canis aureus). Sci. Rep. 13, 7388 (2023).
Darwin, C. The Variation of Animals and Plants Under Domestication Vol. 1 (John Murray, 1868).
Sterndale, R. A. Natural History of the Mammalia of India and Ceylon (Thacker, 1884).
Zimen, E. Der Hund: Abstammung–Verhalten–Mensch und Hund. (Goldmann, 2010).
Rosenberg, S. (BBC, 2002).
Viegas, J. (Animal Planet News, 2002).
Shameer, T., Bhavana, P., Mohan, G., Easa, P. & Sanil, R. Camera traps reveal coat colour variation in an isolated population of golden jackals. Curr. Sci. 122, 738–741 (2022).
Galov, A. et al. First evidence of hybridization between golden jackal (Canis aureus) and domestic dog (Canis familiaris) as revealed by genetic markers. R. Soc. Open Sci. 1, 150450 (2015).
Marklund, L., Moller, M. J., Sandberg, K. & Andersson, L. A missense mutation in the gene for melanocyte-stimulating hormone receptor (MCIR) is associated with the chestnut coat color in horses. Mamm. Genome 7, 895–899 (1996).
Ritland, K., Newton, C. & Marshall, H. D. Inheritance and population structure of the white-phased “Kermode” black bear. Curr. Biol. 11, 1468–1472 (2001).
Peters, L. et al. Born blonde: a recessive loss-of-function mutation in the melanocortin 1 receptor is associated with cream coat coloration in Antarctic fur seals. Ecol. Evol. 6, 5705–5717 (2016).
Newton, J. M. et al. Melanocortin 1 receptor variation in the domestic dog. Mamm. Genome 11, 24–30 (2000).
Brockerville, R. M., McGrath, M. J., Pilgrim, B. L. & Marshall, H. D. Sequence analysis of three pigmentation genes in the Newfoundland population of Canis latrans links the Golden Retriever Mc1r variant to white coat color in coyotes. Mamm. Genome 24, 134–141 (2013).
Schmutz, S. M., Berryere, T. G., Ellinwood, N. M., Kerns, J. A. & Barsh, G. S. MC1R studies in dogs with melanistic mask or brindle patterns. J. Hered. 94, 69–73 (2003).
Lande, R. Mutation and conservation. Conserv. Biol. 9, 782–791 (1995).
Dreger, D. L. & Schmutz, S. M. A new mutation in MC1R explains a coat color phenotype in 2 “old” breeds: Saluki and Afghan hound. J. Hered. 101, 644–649 (2010).
Dürig, N. et al. Two MC1R loss-of-function alleles in cream-coloured Australian cattle dogs and white huskies. Anim. Genet. 49, 284–290 (2018).
Leonard, B. C. et al. Activity, expression and genetic variation of canine β-defensin 103: A multifunctional antimicrobial peptide in the skin of domestic dogs. J. Innate Immun. 4, 248–259 (2012).
Candille, S. I. et al. A defensin mutation causes black coat color in domestic dogs. Science 318, 1418–1423 (2007).
Apollonio, M., Mattioli, L. & Scandura, M. Occurrence of black wolves in the Northern Apennines, Italy. Acta Theriol. 49, 281–285 (2004).
Mowry, C. B., Wilson, L. A. & von Holdt, B. M. Interface of human/wildlife interactions: An example of a Bold Coyote (Canis latrans) in Atlanta, GA, USA. Diversity 13, 372 (2021).
Clark, L. A., Wahl, J. M., Rees, C. A. & Murphy, K. E. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. USA 103, 1376–1381 (2006).
Varga, L. et al. Being Merle: The molecular genetic background of the canine merle mutation. Genes 11, 660 (2020).
Sorsby, A. & Davey, J. B. Ocular associations of dappling (or merling) in the coat colour of dogs. J. Genet. 52, 425–440 (1954).
Langevin, M., Synkova, H., Jancuskova, T. & Pekova, S. Merle phenotypes in dogs: SILV SINE insertions from Mc to Mh. PLoS ONE 13, e0198536 (2018).
Zachos, F. E. et al. Genetic variability, differentiation, and founder effect in golden jackals (Canis aureus) from Serbia as revealed by mitochondrial DNA and nuclear microsatellite loci. Biochem. Genet. 47, 241–250 (2009).
Wang, G.-D. et al. Signature of balancing selection at the MC1R gene in Kunming dog populations. PLoS ONE 8, e55469 (2013).
Ollivier, M. et al. Evidence of coat color variation sheds new light on ancient canids. PLoS ONE 8, e75110 (2013).
Strain, G. M., Clark, L. A., Wahl, J. M., Turner, A. E. & Murphy, K. E. Prevalence of deafness in dogs heterozygous or homozygous for the merle allele. J. Vet. Intern. Med. 23, 282–286 (2009).
Donner, J. et al. Frequency and distribution of 152 genetic disease variants in over 100,000 mixed breed and purebred dogs. PLoS Genet. 14, e1007361 (2018).
Stronen, A. V. et al. Wolf-dog admixture highlights the need for methodological standards and multidisciplinary cooperation for effective governance of wild x domestic hybrids. Biol. Conserv. 266, 109467 (2022).
Fabbri, E. et al. Genetic structure and expansion of golden jackals (Canis aureus) in the north-western distribution range (Croatia and eastern Italian Alps). Conserv. Genet. 15, 187–199 (2014).
Breen, M. et al. Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Res. 11, 1784–1795 (2001).
Vilà, C. et al. Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc. Biol. Sci. 270, 91–97 (2003).
Dayton, M. et al. Developmental validation of short tandem repeat reagent kit for forensic DNA profiling of canine biological material. Croat. Med. J. 50, 268–285 (2009).
Yan, S. et al. Sex identification of dog by PCR based on the differences in the AMELX and AMELY genes. Anim. Genet. 44, 606–606 (2013).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Earl, D. A. & von Holdt, B. M. Structure harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resourc. 4, 359–361 (2012).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research: An update. Bioinformatics 28, 2537–2539 (2012).
Nielsen, E. E., Bach, L. A. & Kotlicki, P. hybridlab (version 10): A program for generating simulated hybrids from population samples. Mol. Ecol. Not. 6, 971–973 (2006).
Jones, O. R. & Wang, J. Colony:A program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resourc. 10, 551–555 (2010).
Kalinowski, S. T., Taper, M. L. & Marshall, T. C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16, 1099–1106 (2007).
Acknowledgements
We are grateful to the colleagues of SEFAG Ltd and Fauna and Flora Foundation, László Kozma, Tamás Trapp, Zsolt Váraljai, and the Institute for Wildlife Management and Nature Conservation of the Hungarian University of Agriculture and Life Sciences for providing golden jackal samples and participating in sample collection, and we are thankful to the University of Veterinary Medicine and private dog owners for providing samples from dogs. The research was supported by the ÚNKP-20-3-II-SZIE-7 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund. This study was financed by the Doctoral School of Animal Biotechnology and Animal Science of the Hungarian University of Agriculture and Life Sciences.
Author information
Authors and Affiliations
Contributions
N.N., F.P.A. and S.V. conceived the research. N.N. and F.P.A. extracted DNA, performed PCR and analysed microsatellite and sequence results. N.N. performed population genetics and kinship analyses. C.E., K.P., H.M., H.M. provided samples and pictures for the research. All authors contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ninausz, N., Fehér, P., Csányi, E. et al. White and other fur colourations and hybridization in golden jackals (Canis aureus) in the Carpathian basin. Sci Rep 13, 21969 (2023). https://doi.org/10.1038/s41598-023-49265-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-49265-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
