
Abstract
The discovery of novel bioactive compounds produced by microorganisms holds significant potential for the development of therapeutics and agrochemicals. In this study, we conducted genome mining to explore the biosynthetic potential of entomopathogenic bacteria belonging to the genera Xenorhabdus and Photorhabdus. By utilizing next-generation sequencing and bioinformatics tools, we identified novel biosynthetic gene clusters (BGCs) in the genomes of the bacteria, specifically plu00736 and plu00747. These clusters were identified as unidentified non-ribosomal peptide synthetase (NRPS) and unidentified type I polyketide synthase (T1PKS) clusters. These BGCs exhibited unique genetic architecture and encoded several putative enzymes and regulatory elements, suggesting its involvement in the synthesis of bioactive secondary metabolites. Furthermore, comparative genome analysis revealed that these BGCs were distinct from previously characterized gene clusters, indicating the potential for the production of novel compounds. Our findings highlighted the importance of genome mining as a powerful approach for the discovery of biosynthetic gene clusters and the identification of novel bioactive compounds. Further investigations involving expression studies and functional characterization of the identified BGCs will provide valuable insights into the biosynthesis and potential applications of these bioactive compounds.
Similar content being viewed by others

Introduction
Microorganisms, particularly bacteria and fungi, have played a crucial role in the discovery and development of numerous drugs used in human medicine1. Approximately 70–80% of important antibiotics currently in use are derived from microorganisms2. For instance, penicillin, streptomycin, tetracycline, erythromycin, and vancomycin, were initially discovered from microorganisms. Furthermore, microorganisms have been instrumental in the discovery of other types of drugs beyond antibiotics. For example, the immunosuppressant drug cyclosporine, the antifungal agent amphotericin B, the antiviral drug acyclovir, and the antiparasitic drug ivermectin are other notable examples also derived from microorganisms3,4,5,6.
In recent years, there has been growing interest in exploring microorganisms, including bacteria and fungi as potential sources of novel drug candidates. Among the bacteria, Xenorhabdus and Photorhabdus bacteria have been valuable sources for the discovery of novel antibiotics and other bioactive compounds. These bacteria, in association with entomopathogenic nematodes, produce a wide range of natural products. For instance, Xenorhabdus spp. produce xenematides7 and xenorxides8, which have proven effective against several pathogenic bacteria. Similarly, Photorhabdus spp. are recognized for their production of multiple antibiotics, including xenorhabdins—a class of toxic macrolide compounds that exhibit robust antimicrobial properties against both Gram-positive and Gram-negative bacteria7,9.
The discovery process often involves screening extracts or purified compounds from these bacteria against panels of bacteria and fungi, including drug-resistant strains. However, advanced techniques like genome sequencing have recently demonstrated that the chemical diversity captured by the traditional culture-based approaches is only the tip of the iceberg, and there are still a significant number of silent or cryptic biosynthetic gene clusters (BGCs), which are not expressed under standard laboratory conditions. Therefore, genome sequencing can be a powerful tool to uncover potential antibiotic candidates, understand the genetic basis of antibiotic production, and guide the discovery and development of novel antibiotics. In this study, we focused on observing BGCs and comparing the of Xenorhabdus and Photorhabdus bacterial genomes from Thailand. Then we can pinpoint genetic variations and potential candidates responsible for novel antibiotic synthesis.
Results
Genome mining of secondary metabolites and BGCs distribution
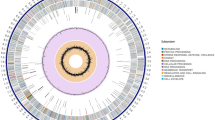
Combining genetic and biosynthetic diversities may provide insight into how these entomopathogenic bacteria can be prioritized in order to uncover novel chemotypes without redundancy of examinations. The processes were initiated with pan-genome analysis, which illustrated the complete set of genes, containing sequences shared by all individuals (core genes) and those that were either shared among specific individuals (accessory genes) or unique to them (singleton genes). The results of this analysis revealed a total of 51,883 genes across the 13 genomes of Xenorhabdus and Photorhabdus bacteria (Supplementary Table S1), covering 10,821 gene clusters, which comprised of 1763 core genes, 5033 accessory genes, and 4024 singleton genes. This step help researchers understand the genetic diversity, evolutionary relationships, and functional capabilities of the strains within the group. Likewise, within the cluster of orthologous groups of proteins (COG20) category, there were a total of 6381 known clusters and 4440 unknown clusters. The COG20 function analysis revealed 6383 known clusters and 4438 unknown clusters, while the COG20 pathway analysis identified 1354 known clusters and 9467 unknown clusters (Fig. 1; Supplementary Table S2, and Supplementary Table S3). Subsequently, an antiSMASH software was employed for the preliminary assessment of the bacterial BGCs. The results shown a total of 314 BGCs across 13 genomes of Xenorhabdus and Photorhabdus bacteria (Supplementary Table S4). The highest biosynthetic diversities were found in X. indica strain KK26.2 (Xind KK26.2) and X. vietnamensis strain NN167.3 (Xvie NN167.3) followed by P. temperata strain MW27.4 (Ptem MW27.4), P. akhurstii strain NN168.5 (Pak NN168.5), P. hainanensis strain NN169.4 (Phai NN169.4), P. laumondii strain MH8.4 (Plau MH8.4), X. miraniensis strain MH16.1 (Xmir MH16.1), and X. ehlersii strain MH9.2 (Xehl MH9.2). The lowest diversity of BGCs was P. australis strain SBR15.4 (Paus SBR15.4), X. stockiae strain RT25.5 (Xsto RT25.5), X. stockiae strain SBRx11.1 (Xsto SBRx11.1), X. japonica strain MW12.3 (Xjap MW12.3), and X. stockiae strain SBR31.4 (Xsto SBR31.4). According to the findings above, the average genome size and BGCs abundance of these entomopathogenic bacteria were 4.6 Mb and 24 BGCs, respectively (Table 1). The standard deviations for these values were calculated to be 562,733.6929 and 3.789323734.

Pan-genome analysis with an average nucleotide identity layer between the bacterial strains. The circular dendrogram was constructed based on the presence or absence of gene clusters. At the upper right corner, average nucleotide identity heatmap are displayed. Each circle represents an individual genome, encompassing all genes (black) from the 13 bacterial genomes. Moving from the outermost layer to inner layers, the core genes (green), accessory genes (blue), and singleton genes (red) were shown. Other information included in the figure comprises the number of contributing genomes, the number of genes in the GC (gene cluster), maximum number of paralogs, geometric homogeneity index, function homogeneity index, combined homogeneity index, Species Core Genome (SCG) clusters, cluster of orthologous groups (COG20) categories, COG20 function, and COG20 pathway.
Sequence-based similarity network of BGCs
Although the program antiSMASH is the most employed bioinformatic tool for finding and evaluating BGCs in the genome sequences10, it is important to note that the structural information of natural products from antiSMASH is occasionally insufficient. Whilst the technique may assess the substrate specificities of NRPS (Non-ribosomal peptide synthetase) or PKS (Polyketide synthase) modules, it does not take nonlinearity, module-skipping, cyclization, or alterations into account. Despite this, the output from antiSMASH and its embedded tools contains an extensive data for structural information11. To assess more accurate biosynthetic abundance, we conducted a network analysis using a combination of bioinformatics tools, including antiSMASH, BiG-SCAPE (Biosynthetic Genes Similarity Clustering and Prospecting Engine)12, and manual verification with our in-house database. Here we reported evidence of a relatively high abundance of 178 putative BGCs from 13 genomes of Xenorhabdus and Photorhabdus bacteria including 89 NRPS, 9 PKS, 22 hybrids, 6 Terpenes, 15 RiPPs (Ribosomally synthesized and post-translationally modified peptides), and 37 others as depicted in Fig. 2 and Supplementary Table S5. Out of the 178 potential biosynthetic clusters, 10 clusters were excluded from the network analysis because insufficient information from the predicted modules. Among the remaining clusters, 146 clusters shown similarity with known BGCs, while 22 clusters represented orphan BGCs for which no known homologous gene clusters could be identified. This suggests the potential novelty of the metabolites associated with these clusters.

Sequence-based similarity network of biosynthetic gene cluster (BGCs) across 13 genomes of Xenorhabdus and Photorhabdus bacteria. The network illustrates identified BGCs, denoted by circular shapes for known BGCs and triangular shapes for unknown BGCs. These clusters are categorized by type, including NRPS (green), PKS (yellow), hybrid (orange), RiPPs (gray), terpene (purple), and others (blue).
Comparison of the biosynthetic gene clusters
In order to identify distinctive or divergent gene clusters associated with potential antimicrobial synthesis, a comparative analysis of the strains' biosynthetic potential was conducted in relation to other strains. The findings revealed that NRPS constituted the predominant class of biosynthetic gene clusters (BGCs) in both Xenorhabdus and Photorhabdus genomes, comprising 51% of the total BGCs. While the ‘others’ and the hybrid groups displayed the second and third highest levels of enrichment and distribution, respectively. In contrast, the PKS, RiPPs, and terpene were comparatively scarce across all the genomes. Through in-silico analysis, it was observed that certain clusters were shared among multiple strains. The clusters exhibited the highest degree of sharing were those resembling known clusters responsible for betalactone production13, followed by gameXpeptides (Gxps) clusters14,15, and clusters associated with photoxenobactin16. Despite the extensive diversity of BGCs in all genomes, only a limited number of clusters associated with secondary metabolite production, such as acinetobactin, ATred, butyrolactone, cuidadopeptide, peptide antimicrobial-Xenorhabdus (PAX peptide), and rhabdopeptide/xenortide peptides (RXPs), were more prevalent only in X. stockiae genomes. Additionally, exclusive discovery of althiomycin, andrimid, and malonomycin clusters was found in X. indica strain Xind KK26.2, P. temperata strain Ptem MW27.4, and P. akhurstii strain Pak NN168.5, respectively (Fig. 3). These strains may hold the potential to serve as distinctive sources for experimental natural product discovery.

A comparative analysis of the biosynthetic gene cluster (BGCs). Different colors in the circles indicate the number of clusters: white for no cluster, gray for 1 cluster, orange for 2 clusters, and red for more than 3 clusters.
Orphan NRPS/PKS gene clusters
Through the analysis of the sequence-based similarity network of BGCs, a total of 22 unknown clusters were discovered. These clusters were further characterized and classified into ten putative BGCs. Among them, three clusters were classified as NRPS clusters, two cluster belonged to the type I T1PKS category, two clusters exhibited hybrid NRPS/PKS characteristics, and three clusters were in ‘others’ group. The nrpks-1 gene clusters were found in the region 25.1 of P. temperata strain Ptem MW27.4, region 3.1 of P. hainanensis strain Phai NN169.4, and region 24.1 of P. akhurstii strain Pak NN168.5 (Fig. 4A). These clusters spanned approximately 37 kb and comprised over 30 genes associated with the biosynthetic process. However, the clusters contained only a single module and showed low sequence similarities to characterized NRPS. Therefore, we were not able to predict the metabolites. The nrpk-2 gene clusters were identified in region 3.1 of both X. stockiae strain Xsto RT25.5 and X. stockiae strain Xsto SBRx11.1 (Fig. 4B). Although, these clusters were not fully sequenced and did not yield any matches in the databases, the clusters consisted of two modules, indicating that the products were likely to be dipeptides, specifically hydrophobic-aliphatic peptides containing valine (Val)—valine (Val) as the constituent building blocks.

The summary of the domain composition and organization of the uncharacterized clusters. The clusters including nrps-1 gene cluster (A), nrps-2 gene cluster (B), t1pks-1and nrps-2 gene cluster (C), t1pks-2 gene cluster (D), nrps-like/pks-1 gene cluster (E) and its presumable compound (F), and nrps-like/ transAT-PKS-like-2 gene cluster (G) and its presumable compound (H).
Within region 8.1 of the X. stockiae strain Xsto RT25.5 and X. stockiae strain Xsto SBRx11.1 genomes, we uncovered one unidentified t1pks-1 gene clusters and one unidentified nrpks-3 gene cluster (Fig. 4C). To distinguish these clusters, we have named them as “plu00736” and “plu00747,” respectively. The absence of matches in the MIBiG, Known ClusterBlast, and our in-house databases emphasized their uniqueness and implied the presence of two potential novel compounds in this genomic region. Moreover, the existence of a self-resistant gene underscored the significance of these genes and suggested their potential antimicrobial activity. Within the t1pks-1 gene clusters, we hypothesized the presence of PKS_KS (Modular-KS), PKS_AT (Modular-AT), PKS_DH Modular-DH), and PKS_KR (Modular-KR) domains which having methylmalonyl-CoA as the substrate. In the NRPS cluster, the genes are predicted to encode 5 modules showing valine (Val)—leucine (Leu)—leucine (Leu)—leucine (Leu)—leucine (Leu) as the primary amino acids. In addition, the t1pks-2 gene clusters, located in region 64.1 of P. akhurstii strain Pak NN168.5 and region 49.1 of P. hainanensis strain Phai NN169.4, displayed a similarity score of 81.37% with t1pks-1 from the region 8.1 of X. stockiae, except for the phosphopantetheine-binding protein (PP-binding) domain. These clusters could potentially represent analogues of the same compound (Fig. 4D).
Regarding the hybrid-1 gene clusters from nrps-like and t1pks-1 genes in the region 16.1 of X. stockiae strain Xsto RT25.5, region 16.1 of X. stockiae strain Xsto SBRx11.1, and region 11.1 of X. stockiae strain Xsto SBR31.4 spanned 57.4 kb in size and contained up to forty related genes, whose assembly line was composed of seven modules (Fig. 4E). According to the assembly line rule and the substrates of domains, the gene clusters were assumed to synthesize the product shown in Fig. 4F. From NORINE database, the structure has similar characteristics to those of a bacterial small molecule that is used worldwide as a protease inhibitor. This molecule has served as a well-established chemical model in the fields of autophagy and immunoproteasome research17. Hybrid-2 gene clusters is a NRPS-like/transAT-PKS-like hybrid gene in X. miraniensis strain Xmir MH16.1 region 32.1, X. vietnamensis strain Xvei NN167.3 region 42.1, and X. japonica strain Xjap MW12.3 region 30.1 encoding a protein comprising five modules and one domain was predicted to incorporate cysteine (Cys) as the substrates (Fig. 4G); therefore, the products were predicted to be hexapeptides including one cysteine molecules (Fig. 4H). For the remaining clusters (data not shown), three distinct types were identified. Firstly, in the X. miraniensis strain Xmir MH16.1 region 23.1 and X. indica strain Xind KK26.2 region 22, we observed nucleotide-related clusters. In X. japonica strain Xjap MW12.3 region 43.1 and X. indica strain Xind KK26.2 region 14.1, phosphonate-related clusters were presented and within the X. miraniensis strain Xmir MH16.1 region 32.1, we identified lanthipeptide-class-II clusters. Unfortunately, none of these modules could be predicted from the gene clusters, and they exhibited low sequence similarities to characterized compounds. As a result, we were unable to predict the metabolites associated with these clusters.
Discussion
Genome mining of secondary metabolites and BGCs distribution
The average size of the 13 Xenorhabdus and Photorhabdus genomes from Thailand was approximately 4.6 Mb with an average of 24 biosynthetic gene clusters (BGCs). The count of BGCs is deemed high when compared to other genera in the same family18. A greater number of BGCs indicated a greater potential to produce bioactive substances and secondary metabolites19. Furthermore, the outcomes from analyzing the pan-genome revealed certain resemblances in the genomic sequences of these two bacterial genera. Both bacteria have a set of core genes which are commonly found across species. These core genes are essential for basic cellular functions and are relatively conserved in terms of sequence and function20. Nevertheless, there are noteworthy distinctions observed between the two genera. Each genus harbors a unique collection of genes specific to its genus and species, which play a pivotal role in shaping their distinct characteristics and lifestyles13. These genes reflect their adaptation to diverse ecological niches, encompassing regulatory elements as well as the production of secondary metabolites such as antibiotics and toxins, which are uncommonly encountered in other organisms21. Likewise, the genes associated with the biosynthesis of secondary metabolites can demonstrate considerable variability and diversity among different species and strains. Such variations play a significant role in generating a wide array of distinct bioactive compounds.
Sequence-based similarity network of BGCs
The analysis highlighted the diversity of BGCs within the genus, indicating the biosynthetic potential of these symbiotic bacterium, including the possibility of biodiscovery of novel secondary compounds from the isolates. A total of 160 biosynthetic clusters with likeness to recognized BGCs contribute to the bacteria's role in pathogenesis, defense against competing microorganisms, and the establishment of the nematode-bacteria symbiosis which has been found to exhibit significant inhibitory effects against various microorganisms, including bacteria, fungi, and some parasites7,9,22. Among the strains examined in this study, X. indica strain Xind KK26.2 and X. vietnamensis strain Xvei NN167.3 prominently exhibit the highest prevalence of the secondary metabolite biosynthetic gene clusters (smBGCs), suggesting their potential for uncovering novel bioactive secondary metabolites. This hypothesis is further supported by antiSMASH analyses, revealing that both X. indica strain Xind KK26.2 and X. vietnamensis strain Xvei NN167.3 strains possess an enrichment of over 28 BGCs. As a result, they are attractive candidates for the development of novel antibiotics or antimicrobial medicines. However, additional research is required to completely comprehend its prospective uses and enhance its effectiveness. During the subsequent analysis, the refined genomes exhibited similarities by harboring gene clusters associated with known clusters including xenocoumacin23,24,25, szentirazine26, frederiksenibactin27, photoxenobactin16, tilivalline28, putrebactin29, odilorhabdin30, o-antigen31, gameXpeptides (Gxps)14,15, mevalagmapeptide15, betalactone13, kolossin32, isopropylstilbene (IPS)33, CDPS34, malonomycin35, acinetobactin36, aryl polyene37, ririwpeptide15, andrimid38,39, pyrrolizixenamide40, xenoamicin41, xenorhabdin42, lipocitides13, fabclavine43, Acyltransferase Red (ATred)44, phenazine45, cuidadopeptide26,46, althiomycin47, PAX (peptide-antimicrobial-Xenorhabdus)48,49,50, xenematide7, nematophin51, rhabdopeptide (RXPs)52, glidobactin53, and butyrolactone54.
Comparison of the biosynthetic gene clusters
The findings of this study exhibited remarkable similarities to the analysis conducted by Shi and colleagues in 2022, where NRPS were identified as the most abundant class of biosynthetic gene clusters (BGCs) in both Xenorhabdus and Photorhabdus genomes. The high prevalence of NRPS BGCs suggests that their products could play significant ecological roles13. Additionally, the others group emerged as the second-largest class of BGCs, with its products potentially aiding bacteria in performing specific ecological functions13. The hybrid class of PKS/NRPS shows a slightly enrichment and distribution, while PKS, RiPPs, and terpene BGCs are relatively scarce across all genomes compared to other types. Moreover, in alignment with previous studies13, betalactone clusters were identified as the predominant cluster type followed by gameXpeptides and photoxenobactin biosynthetic clusters. The products of betalactone and Gxps clusters have been identified to play a role in insect immune suppression, while photoxenobactin has been found to exhibit insecticidal properties13. In addition to the mentioned clusters, the remaining clusters displayed similarities to various types of bioactive compounds with antibacterial, insecticidal, antiprotozoal, antifungal, antiparasitic properties, as well as broad-spectrum compounds like fabclavine43.
Orphan NRPS/PKS gene clusters
With regards to the orphan clusters, all clusters except the T1PSK type were found to encompass genes associated with transcription regulation and transport. Previous studies have emphasized the significance of these transcription regulation and transport mechanisms in the biosynthesis of antibiotics like oleandomycin55 and spiramycin56. By integrating comparative genomics with sequence-based similarity network analyses, researchers have gained valuable insights into the genetic and BGCs (biosynthetic gene clusters) diversity present within and between Xenorhabdus spp. and Photorhabdus spp. These findings contribute to our comprehension of the distinctive or divergent gene clusters associated with their potential capacity for antimicrobial synthesis.
Methods
Entomopathogenic bacterial strains
Thirteen isolates of symbiotic bacteria were isolated from entomopathogenic nematodes from Thailand. Eight isolates of Xenorhabdus and 5 isolates of Photorhabdus were previously identified with recA sequence57,58,59,60,61,62 (Table 2).
Genome sequencing and annotation
To initiate the experiment, a single colony of each strain was inoculated in 5 ml of Luria–Bertani broth40 and incubated at 28 °C with agitation overnight. Genomic DNA was isolated using the DNeasy kit (Qiagen, Hilden, Germany). For next-generation sequencing, mate pair libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer's instructions. All libraries underwent sequencing in 250 bp paired read runs on the Illumina MiSeq platform. The resulting reads were subjected to quality trimming using Sickle v.1.3363, discarding any trimmed reads shorter than 125 bp. Subsequently, genome assembly was performed using SPAdes v. 3.10.164 with the following parameters: --cov-cutoff auto, --careful in paired-end mode plus mate pairs, and k-mer lengths of 21, 33, 55, 77, 81, and 91. Genome annotation was conducted using Prokka v. 1.1265 with the following parameters: --usegenus --genus GENUS –addgenes --evalue 0.0001 --rfam --kingdom Bacteria --gcode 11 --gram --mincontiglen 200.
Bioinformatics analysis
The pan and core genome analysis of annotated genomes herein mainly followed the anvi’o 7.1 pan genomic workflow66,67. Briefly, an anvi’o genomes-storage-db was generated using the program anvi-gen-genomes-storage. After that, an anvi’o pan-db was performed using the program anvi-pan-genome. Lastly, the results were displayed in anvi’o interactive interface using the program anvi-display-pan67. After that, the 13 genome sequences, annotated in the GenBank format, were analyzed for biosynthetic gene cluster (BGC) identification using the antiSMASH (antibiotics and secondary metabolite analysis shell) 6.1.1 pipeline. The detection strictness was set to “relaxed,” and the ClusterBlast, Cluster Pfam analysis, and Pfam-based GO options were enabled for the antiSMASH analysis. After the initial BGC types were assigned by antiSMASH, a manual inspection and reclassification process took place, ensuring accurate classification. A summary of the annotated BGCs was then generated. Subsequently, the antiSMASH job IDs were utilized to explore and classify the BGCs using the biosynthetic gene cluster family's database, BiG-FAM 1.0.068, providing preliminary insights. To refine the analysis, BiG-SCAPE 1.0.068 was employed with a cutoff value of 0.65. The resulting gene cluster families (GCFs) underwent a thorough manual double-check using an interactive network, and necessary corrections were applied. Finally, the conclusive outcomes were visualized as a network using Cytoscape 3.10.
Data availability
The genome sequence of Xenorhabdus and Photorhabdus data that support the findings of this study are available in NCBI GenBank database under accession SAMN36278238 to SAMN36278250. All other data generated or analysed in this study are available within the article and its supplementary information. Supporting Information files are published exactly as provided, and are not modified or copyedited.
References
Katz, L. & Baltz, R. H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 43, 155–176 (2016).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661 (2016).
Brautaset, T. et al. Biosynthesis of the polyene antifungal antibiotic nystatin in Streptomyces noursei ATCC 11455: Analysis of the gene cluster and deduction of the biosynthetic pathway. Chem. Biol. 7, 395–403 (2000).
Liu, X. F. et al. Actinomycin D enhances killing of cancer cells by immunotoxin RG7787 through activation of the extrinsic pathway of apoptosis. Proc. Natl. Acad. Sci. 113, 10666–10671 (2016).
Schwecke, T. et al. The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proc. Natl. Acad. Sci. 92, 7839–7843 (1995).
Waldron, C. et al. Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa1. Chem. Biol. 8, 487–499 (2001).
Tobias, N. J. et al. Natural product diversity associated with the nematode symbionts Photorhabdus and Xenorhabdus. Nat. Microbiol. 2, 1676–1685 (2017).
Webster, J. M., Li, J. & Chen, G. Xenorxides with antibacterial and antimycotic properties. Google Patents. https://patents.google.com/patent/WO1996032396A1/en (1996).
Bode, E. et al. Biosynthesis and function of simple amides in Xenorhabdus doucetiae. Environ. Microbiol. 19, 4564–4575 (2017).
Blin, K. et al. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87 (2019).
Tietz, J. & Mitchell, A. Using genomics for natural product structure elucidation. Curr. Top. Med. Chem. 16, 1645–1694 (2016).
Navarro-Muñoz, J. C. et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68 (2020).
Shi, Y.-M. et al. Global analysis of biosynthetic gene clusters reveals conserved and unique natural products in entomopathogenic nematode-symbiotic bacteria. Nat. Chem. 14, 701–712 (2022).
Fu, J. et al. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 30, 440–446 (2012).
Wang, G. et al. CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat. Microbiol. 4, 2498–2510 (2019).
Shi, Y.-M., Hirschmann, M., Shi, Y.-N. & Bode, H. B. Cleavage off-loading and post-assembly-line conversions yield products with unusual termini during biosynthesis. ACS Chem. Biol. 17, 2221–2228 (2022).
Li, J. H. et al. Making and breaking leupeptin protease inhibitors in pathogenic Gammaproteobacteria. Angew. Chem. Int. Ed. 59, 17872–17880 (2020).
Mohite, O. S., Lloyd, C. J., Monk, J. M., Weber, T. & Palsson, B. O. Pangenome analysis of Enterobacteria reveals richness of secondary metabolite gene clusters and their associated gene sets. Synth. Syst. Biotechnol. 7, 900–910 (2022).
Yamanaka, K. et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. 111, 1957–1962 (2014).
Tettelin, H. et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. 102, 13950–13955 (2005).
Jordan, I. K., Makarova, K. S., Spouge, J. L., Wolf, Y. I. & Koonin, E. V. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 11, 555–565 (2001).
Shi, Y.-M. & Bode, H. B. Chemical language and warfare of bacterial natural products in bacteria–nematode–insect interactions. Nat. Prod. Rep. 35, 309–335 (2018).
Park, D. et al. Genetic analysis of xenocoumacin antibiotic production in the mutualistic bacterium Xenorhabdus nematophila. Mol. Microbiol. 73, 938–949 (2009).
Reimer, D., Luxenburger, E., Brachmann, A. O. & Bode, H. B. A new type of pyrrolidine biosynthesis is involved in the late steps of xenocoumacin production in Xenorhabdus nematophila. ChemBioChem 10, 1997–2001 (2009).
Lang, G., Kalvelage, T., Peters, A., Wiese, J. & Imhoff, J. F. Linear and cyclic peptides from the entomopathogenic bacterium Xenorhabdus nematophilus. J. Nat. Prod. 71, 1074–1077 (2008).
Bode, E. et al. Promoter activation in Δhfq mutants as an efficient tool for specialized metabolite production enabling direct bioactivity testing. Angew. Chem. 131, 19133–19139 (2019).
Stow, P. R., Reitz, Z. L., Johnstone, T. C. & Butler, A. Genomics-driven discovery of chiral triscatechol siderophores with enantiomeric Fe (iii) coordination. Chem. Sci. 12, 12485–12493 (2021).
Dornisch, E. et al. Biosynthesis of the enterotoxic pyrrolobenzodiazepine natural product tilivalline. Angew. Chem. Int. Ed. 56, 14753–14757 (2017).
Hirschmann, M., Grundmann, F. & Bode, H. B. Identification and occurrence of the hydroxamate siderophores aerobactin, putrebactin, avaroferrin and ochrobactin C as virulence factors from entomopathogenic bacteria. Environ. Microbiol. 19, 4080–4090 (2017).
Pantel, L. et al. Odilorhabdins, antibacterial agents that cause miscoding by binding at a new ribosomal site. Mol. Cell 70, 83-94.e87 (2018).
Bélanger, M., Burrows, L. L. & Lam, J. S. Functional analysis of genes responsible for the synthesis of the B-band O antigen of Pseudomonas aeruginosa serotype O6 lipopolysaccharide. Microbiology 145, 3505–3521 (1999).
Bode, H. B. et al. Structure elucidation and activity of kolossin A, the d-/l-pentadecapeptide product of a giant nonribosomal peptide synthetase. Angew. Chem. Int. Ed. 54, 10352–10355 (2015).
Kavakli, S., Grammbitter, G. L. & Bode, H. B. Biosynthesis of the multifunctional isopropylstilbene in Photorhabdus laumondii involves cross-talk between specialized and primary metabolism. Tetrahedron 128, 133116 (2022).
Imai, Y. et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 576, 459–464 (2019).
Law, B. J. et al. A vitamin K-dependent carboxylase orthologue is involved in antibiotic biosynthesis. Nat. Catal. 1, 977–984 (2018).
Mihara, K. et al. Identification and transcriptional organization of a gene cluster involved in biosynthesis and transport of acinetobactin, a siderophore produced by Acinetobacter baumannii ATCC 19606T. Microbiology 150, 2587–2597 (2004).
Grammbitter, G. L. et al. An uncommon type II PKS catalyzes biosynthesis of aryl polyene pigments. J. Am. Chem. Soc. 141, 16615–16623 (2019).
Liu, X., Fortin, P. D. & Walsh, C. T. Andrimid producers encode an acetyl-CoA carboxyltransferase subunit resistant to the action of the antibiotic. Proc. Natl. Acad. Sci. 105, 13321–13326 (2008).
Ratnayake, N. D., Wanninayake, U., Geiger, J. H. & Walker, K. D. Stereochemistry and mechanism of a microbial phenylalanine aminomutase. J. Am. Chem. Soc. 133, 8531–8533 (2011).
Schimming, O. et al. Structure, biosynthesis, and occurrence of bacterial pyrrolizidine alkaloids. Angew. Chem. Int. Ed. 54, 12702–12705 (2015).
Zhou, Q. et al. Structure and biosynthesis of xenoamicins from entomopathogenic Xenorhabdus. Chemistry 19, 16772–16779 (2013).
Walia, S., Sharma, K. & Ganguli, S. Entomopathogenic nematodebacterium complex derived novel antibiotics and their pest control properties. In Proceedings of Short Term National Training Course entitled “Advanced Techniques for Exploiting the ENBI Complexes (Entomopathogenic Nematodes-bacterial symbionts and the Insect hosts) for Biomanagement of Insect Pests of Crops (Indian Agricultural Research Institute, 2011).
Wenski, S. L., Kolbert, D., Grammbitter, G. L. & Bode, H. B. Fabclavine biosynthesis in X. szentirmaii: Shortened derivatives and characterization of the thioester reductase FclG and the condensation domain-like protein FclL. J. Ind. Microbiol. Biotechnol. 46, 565–572 (2019).
Tietze, A., Shi, Y. N., Kronenwerth, M. & Bode, H. B. Nonribosomal peptides produced by minimal and engineered synthetases with terminal reductase domains. ChemBioChem 21, 2750–2754 (2020).
Laursen, J. B. & Nielsen, J. Phenazine natural products: Biosynthesis, synthetic analogues, and biological activity. Chem. Rev. 104, 1663–1686 (2004).
Dudnik, A., Bigler, L. & Dudler, R. Heterologous expression of a Photorhabdus luminescens syrbactin-like gene cluster results in production of the potent proteasome inhibitor glidobactin A. Microbiol. Res. 168, 73–76 (2013).
Gerc, A. J., Song, L., Challis, G. L., Stanley-Wall, N. R. & Coulthurst, S. J. The insect pathogen Serratia marcescens Db10 uses a hybrid non-ribosomal peptide synthetase-polyketide synthase to produce the antibiotic althiomycin. PLoS One 7, e44673 (2012).
Dreyer, J. et al. Xenorhabdus khoisanae SB10 produces Lys-rich PAX lipopeptides and a Xenocoumacin in its antimicrobial complex. BMC Microbiol. 19, 1–11 (2019).
Fuchs, S. W., Proschak, A., Jaskolla, T. W., Karas, M. & Bode, H. B. Structure elucidation and biosynthesis of lysine-rich cyclic peptides in Xenorhabdus nematophila. Org. Biomol. Chem. 9, 3130–3132 (2011).
Gualtieri, M., Aumelas, A. & Thaler, J.-O. Identification of a new antimicrobial lysine-rich cyclolipopeptide family from Xenorhabdus nematophila. J. Antibiot. 62, 295–302 (2009).
Cai, X. et al. Biosynthesis of the antibiotic nematophin and its elongated derivatives in entomopathogenic bacteria. Org. Lett. 19, 806–809 (2017).
Reimer, D. et al. Rhabdopeptides as insect-specific virulence factors from entomopathogenic bacteria. ChemBioChem 14, 1991–1997 (2013).
Bian, X., Plaza, A., Zhang, Y. & Müller, R. Luminmycins A-C, cryptic natural products from Photorhabdus luminescens identified by heterologous expression in Escherichia coli. J. Nat. Prod. 75, 1652–1655 (2012).
Guo, C. J. et al. Application of an efficient gene targeting system linking secondary metabolites to their biosynthetic genes in Aspergillus terreus. Org. Lett. 15, 3562–3565 (2013).
Dyson, P. Streptomyces. In Encyclopedia of Microbiology 3rd edn (ed. Schaechter, M.) 318–332 (Academic Press, 2009).
Nguyen, H. C. et al. Glycosylation steps during spiramycin biosynthesis in Streptomyces ambofaciens: Involvement of three glycosyltransferases and their interplay with two auxiliary proteins. Antimicrob. Agents Chemother. 54, 2830–2839 (2010).
Fukruksa, C. et al. Isolation and identification of Xenorhabdus and Photorhabdus bacteria associated with entomopathogenic nematodes and their larvicidal activity against Aedes aegypti. Parasites Vectors 10, 1–10 (2017).
Muangpat, P. et al. Antibacterial activity of Xenorhabdus and Photorhabdus isolated from entomopathogenic nematodes against antibiotic-resistant bacteria. PLoS One 15, e0234129 (2020).
Muangpat, P. et al. Screening of the antimicrobial activity against drug resistant bacteria of Photorhabdus and Xenorhabdus associated with entomopathogenic nematodes from Mae Wong National Park, Thailand. Front. Microbiol. 8, 1142 (2017).
Yimthin, T. et al. A study on Xenorhabdus and Photorhabdus isolates from Northeastern Thailand: Identification, antibacterial activity, and association with entomopathogenic nematode hosts. PLoS One 16, e0255943 (2021).
Yooyangket, T. et al. Identification of entomopathogenic nematodes and symbiotic bacteria from Nam Nao National Park in Thailand and larvicidal activity of symbiotic bacteria against Aedes aegypti and Aedes albopictus. PLoS One 13, e0195681 (2018).
Thanwisai, A. et al. Diversity of Xenorhabdus and Photorhabdus spp. and their symbiotic entomopathogenic nematodes from Thailand. PLoS One 7, e43835 (2012).
Joshi, N. & Fass, J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. https://github.com/najoshi/sickle (2011).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Delmont, T. O. & Eren, A. M. Linking pangenomes and metagenomes: The Prochlorococcus metapangenome. PeerJ 6, e4320 (2018).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
Kautsar, S. A., Blin, K., Shaw, S., Weber, T. & Medema, M. H. BiG-FAM: The biosynthetic gene cluster families database. Nucleic Acids Res. 49, D490–D497 (2021).
Funding
This work was supported by the Royal Golden Jubilee Ph.D. Program (Grant No: PHD/0084/2561), through the National Research Council of Thailand (former Thailand Research Fund) and National Science, Research and Innovation Fund (Grant No. R2565B050).
Author information
Authors and Affiliations
Contributions
A.T. Conceptualization and supervision of the study; A.T., W.M., Y.-M.S., H.B.B. conceived and designed the experiments and interpretation of data; A.T., W.M., P.M., S.S., and A.V. performed experiments and prepared the figures; A.T., and W.M. funding acquisition and drafted the manuscript; T.R., N.C., R.A.R.M. Revised the manuscript; All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meesil, W., Muangpat, P., Sitthisak, S. et al. Genome mining reveals novel biosynthetic gene clusters in entomopathogenic bacteria. Sci Rep 13, 20764 (2023). https://doi.org/10.1038/s41598-023-47121-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-47121-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
